NF-κB
NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) is a protein complex that controls transcription of DNA, cytokine production and cell survival. NF-κB is found in almost all animal cell types and is involved in cellular responses to stimuli such as stress, cytokines, free radicals, heavy metals, ultraviolet irradiation, oxidized LDL, and bacterial or viral antigens.[1][2][3][5][6] NF-κB plays a key role in regulating the immune response to infection. Incorrect regulation of NF-κB has been linked to cancer, inflammatory and autoimmune diseases, septic shock, viral infection, and improper immune development. NF-κB has also been implicated in processes of synaptic plasticity and memory.[7][8][9][10][11][12]
Discovery
NF-κB was discovered by Ranjan Sen (NIH) in the lab of Nobel laureate David Baltimore via its interaction with an 11-base pair sequence in the immunoglobulin light-chain enhancer in B cells.[13]
Structure
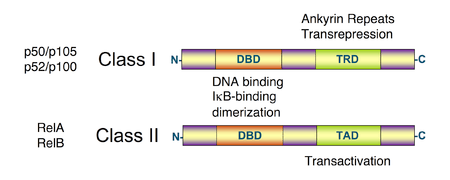
All proteins of the NF-κB family share a Rel homology domain in their N-terminus. A subfamily of NF-κB proteins, including RelA, RelB, and c-Rel, have a transactivation domain in their C-termini. In contrast, the NF-κB1 and NF-κB2 proteins are synthesized as large precursors, p105, and p100, which undergo processing to generate the mature NF-κB subunits, p50 and p52, respectively. The processing of p105 and p100 is mediated by the ubiquitin/proteasome pathway and involves selective degradation of their C-terminal region containing ankyrin repeats. Whereas the generation of p52 from p100 is a tightly regulated process, p50 is produced from constitutive processing of p105.[14][15] The p50 and p52 proteins have no intrinsic ability to activate transcription and thus have been proposed to act as transcriptional repressors when binding κB elements as homodimers.[16][17] Indeed, this confounds the interpretation of p105-knockout studies, where the genetic manipulation is removing an IκB (full-length p105) and a likely repressor (p50 homodimers) in addition to a transcriptional activator (the RelA-p50 heterodimer).
Members
NF-κB family members share structural homology with the retroviral oncoprotein v-Rel, resulting in their classification as NF-κB/Rel proteins.[1]
There are five proteins in the mammalian NF-κB family:[18]
| Class | Protein | Aliases | Gene |
|---|---|---|---|
| I | NF-κB1 | p105 → p50 | NFKB1 |
| NF-κB2 | p100 → p52 | NFKB2 | |
| II | RelA | p65 | RELA |
| RelB | RELB | ||
| c-Rel | REL |
Below are the five human NF-κB family members:
| NFKB1 | |
|---|---|
 | |
| Identifiers | |
| Symbol | NFKB1 |
| NCBI gene | 4790 |
| HGNC | 7794 |
| OMIM | 164011 |
| RefSeq | NM_003998 |
| UniProt | P19838 |
| Other data | |
| Locus | Chr. 4 q24 |
| RELA | |
|---|---|
 | |
| Identifiers | |
| Symbol | RELA |
| NCBI gene | 5970 |
| HGNC | 9955 |
| OMIM | 164014 |
| RefSeq | NM_021975 |
| UniProt | Q04206 |
| Other data | |
| Locus | Chr. 11 q13 |
| NFKB2 | |
|---|---|
| Identifiers | |
| Symbol | NFKB2 |
| NCBI gene | 4791 |
| HGNC | 7795 |
| OMIM | 164012 |
| RefSeq | NM_002502 |
| UniProt | Q00653 |
| Other data | |
| Locus | Chr. 10 q24 |
| RELB | |
|---|---|
| Identifiers | |
| Symbol | RELB |
| NCBI gene | 5971 |
| HGNC | 9956 |
| OMIM | 604758 |
| RefSeq | NM_006509 |
| UniProt | Q01201 |
| Other data | |
| Locus | Chr. 19 q13.2-19q13 |
| REL | |
|---|---|
| Identifiers | |
| Symbol | REL |
| NCBI gene | 5966 |
| HGNC | 9954 |
| OMIM | 164910 |
| RefSeq | NM_002908 |
| UniProt | Q04864 |
| Other data | |
| Locus | Chr. 2 p13-p12 |
Species distribution and evolution
In addition to mammals, NF-κB is found in a number of simple animals as well.[19] These include cnidarians (such as sea anemones, coral and hydra), porifera (sponges), the single-celled eukaryote Capsaspora owczarzaki and insects (such as moths, mosquitoes and fruitflies). The sequencing of the genomes of the mosquitoes A. aegypti and A. gambiae, and the fruitfly D. melanogaster has allowed comparative genetic and evolutionary studies on NF-κB. In those insect species, activation of NF-κB is triggered by the Toll pathway (which evolved independently in insects and mammals) and by the Imd (immune deficiency) pathway.[20]
Signaling
Effect of activation
NF-κB is important in regulating cellular responses because it belongs to the category of "rapid-acting" primary transcription factors, i.e., transcription factors that are present in cells in an inactive state and do not require new protein synthesis in order to become activated (other members of this family include transcription factors such as c-Jun, STATs, and nuclear hormone receptors). This allows NF-κB to be a first responder to harmful cellular stimuli. Known inducers of NF-κB activity are highly variable and include reactive oxygen species (ROS), tumor necrosis factor alpha (TNFα), interleukin 1-beta (IL-1β), bacterial lipopolysaccharides (LPS), isoproterenol, cocaine, and ionizing radiation.[22]
Receptor activator of NF-κB (RANK), which is a type of TNFR, is a central activator of NF-κB. Osteoprotegerin (OPG), which is a decoy receptor homolog for RANK ligand (RANKL), inhibits RANK by binding to RANKL, and, thus, osteoprotegerin is tightly involved in regulating NF-κB activation.[23]
Many bacterial products and stimulation of a wide variety of cell-surface receptors lead to NF-κB activation and fairly rapid changes in gene expression.[1] The identification of Toll-like receptors (TLRs) as specific pattern recognition molecules and the finding that stimulation of TLRs leads to activation of NF-κB improved our understanding of how different pathogens activate NF-κB. For example, studies have identified TLR4 as the receptor for the LPS component of Gram-negative bacteria.[24] TLRs are key regulators of both innate and adaptive immune responses.[25]
Unlike RelA, RelB, and c-Rel, the p50 and p52 NF-κB subunits do not contain transactivation domains in their C terminal halves. Nevertheless, the p50 and p52 NF-κB members play critical roles in modulating the specificity of NF-κB function. Although homodimers of p50 and p52 are, in general, repressors of κB site transcription, both p50 and p52 participate in target gene transactivation by forming heterodimers with RelA, RelB, or c-Rel.[26] In addition, p50 and p52 homodimers also bind to the nuclear protein Bcl-3, and such complexes can function as transcriptional activators.[27][28][29]
Inhibition
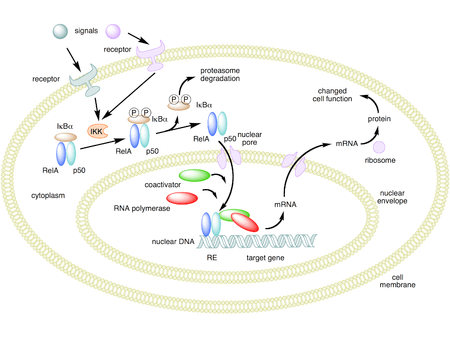
In unstimulated cells, the NF-κB dimers are sequestered in the cytoplasm by a family of inhibitors, called IκBs (Inhibitor of κB), which are proteins that contain multiple copies of a sequence called ankyrin repeats. By virtue of their ankyrin repeat domains, the IκB proteins mask the nuclear localization signals (NLS) of NF-κB proteins and keep them sequestered in an inactive state in the cytoplasm.[30]
IκBs are a family of related proteins that have an N-terminal regulatory domain, followed by six or more ankyrin repeats and a PEST domain near their C terminus. Although the IκB family consists of IκBα, IκBβ, IκBε, and Bcl-3, the best-studied and major IκB protein is IκBα. Due to the presence of ankyrin repeats in their C-terminal halves, p105 and p100 also function as IκB proteins. The c-terminal half of p100, that is often referred to as IκBδ, also functions as an inhibitor.[31][32] IκBδ degradation in response to developmental stimuli, such as those transduced through LTβR, potentiate NF-κB dimer activation in a NIK dependent non-canonical pathway.[31][33]
Activation process (canonical/classical)
Activation of the NF-κB is initiated by the signal-induced degradation of IκB proteins. This occurs primarily via activation of a kinase called the IκB kinase (IKK). IKK is composed of a heterodimer of the catalytic IKKα and IKKβ subunits and a "master" regulatory protein termed NEMO (NF-κB essential modulator) or IKKγ. When activated by signals, usually coming from the outside of the cell, the IκB kinase phosphorylates two serine residues located in an IκB regulatory domain. When phosphorylated on these serines (e.g., serines 32 and 36 in human IκBα), the IκB proteins are modified by a process called ubiquitination, which then leads them to be degraded by a cell structure called the proteasome.
With the degradation of IκB, the NF-κB complex is then freed to enter the nucleus where it can 'turn on' the expression of specific genes that have DNA-binding sites for NF-κB nearby. The activation of these genes by NF-κB then leads to the given physiological response, for example, an inflammatory or immune response, a cell survival response, or cellular proliferation. Translocation of NF-κB to nucleus can be detected immunocytochemically and measured by laser scanning cytometry.[34] NF-κB turns on expression of its own repressor, IκBα. The newly synthesized IκBα then re-inhibits NF-κB and, thus, forms an auto feedback loop, which results in oscillating levels of NF-κB activity.[35] In addition, several viruses, including the AIDS virus HIV, have binding sites for NF-κB that controls the expression of viral genes, which in turn contribute to viral replication or viral pathogenicity. In the case of HIV-1, activation of NF-κB may, at least in part, be involved in activation of the virus from a latent, inactive state.[36] YopP is a factor secreted by Yersinia pestis, the causative agent of plague, that prevents the ubiquitination of IκB. This causes this pathogen to effectively inhibit the NF-κB pathway and thus block the immune response of a human infected with Yersinia.[37]
Inhibitors of NF-κB activity
Concerning known protein inhibitors of NF-κB activity, one of them is IFRD1, which represses the activity of NF-κB p65 by enhancing the HDAC-mediated deacetylation of the p65 subunit at lysine 310, by favoring the recruitment of HDAC3 to p65. In fact IFRD1 forms trimolecular complexes with p65 and HDAC3.[38][39]
The NAD+-dependent protein deacetylase and longevity factor SIRT1 inhibits NF-κB gene expression by deacetylating the RelA/p65 subunit of NF-kB at lysine 310.[40]
Non-canonical/alternate pathway
A select set of cell-differentiating or developmental stimuli, such as lymphotoxin β-receptor (LTβR), BAFF or RANKL, activate the non-canonical NF-κB pathway to induce NF-κB/RelB:p52 dimer in the nucleus. In this pathway, activation of the NF-κB inducing kinase (NIK) upon receptor ligation led to the phosphorylation and subsequent proteasomal processing of the NF-κB2 precursor protein p100 into mature p52 subunit in an IKK1/IKKa dependent manner. Then p52 dimerizes with RelB to appear as a nuclear RelB:p52 DNA binding activity and regulate a distinct class of genes.[41] In contrast to the canonical signaling that relies upon NEMO-IKK2 mediated degradation of IκBα, -β, -ε, the non-canonical signaling critically depends on NIK mediated processing of p100 into p52. Given their distinct regulations, these two pathways were thought to be independent of each other. However, recent analyses revealed that synthesis of the constituents of the non-canonical pathway, viz RelB and p52, is controlled by the canonical IKK2-IκB-RelA:p50 signaling.[42] Moreover, generation of the canonical and non-canonical dimers, viz RelA:p50 and RelB:p52, within the cellular milieu are also mechanistically interlinked.[42] These analyses suggest that an integrated NF-κB system network underlies activation of both RelA and RelB containing dimer and that a malfunctioning canonical pathway will lead to an aberrant cellular response also through the non-canonical pathway.
In immunity
NF-κB is a major transcription factor that regulates genes responsible for both the innate and adaptive immune response.[43] Upon activation of either the T- or B-cell receptor, NF-κB becomes activated through distinct signaling components. Upon ligation of the T-cell receptor, protein kinase Lck is recruited and phosphorylates the ITAMs of the CD3 cytoplasmic tail. ZAP70 is then recruited to the phosphorylated ITAMs and helps recruit LAT and PLC-γ, which causes activation of PKC. Through a cascade of phosphorylation events, the kinase complex is activated and NF-κB is able to enter the nucleus to upregulate genes involved in T-cell development, maturation, and proliferation.[44]
In the nervous system
In addition to roles in mediating cell survival, studies by Mark Mattson and others have shown that NF-κB has diverse functions in the nervous system including roles in plasticity, learning, and memory. In addition to stimuli that activate NF-κB in other tissues, NF-κB in the nervous system can be activated by Growth Factors (BDNF, NGF) and synaptic transmission such as glutamate.[8] These activators of NF-κB in the nervous system all converge upon the IKK complex and the canonical pathway.
Recently there has been a great deal of interest in the role of NF-κB in the nervous system. Current studies suggest that NF-κB is important for learning and memory in multiple organisms including crabs,[10][11] fruit flies,[45] and mice.[8][9] NF-κB may regulate learning and memory in part by modulating synaptic plasticity,[7][46] synapse function,[45][47][48] as well as by regulating the growth of dendrites[49] and dendritic spines.[48]
Genes that have NF-κB binding sites are shown to have increased expression following learning,[9] suggesting that the transcriptional targets of NF-κB in the nervous system are important for plasticity. Many NF-κB target genes that may be important for plasticity and learning include growth factors (BDNF, NGF)[50] cytokines (TNF-alpha, TNFR)[51] and kinases (PKAc).[46]
Despite the functional evidence for a role for Rel-family transcription factors in the nervous system, it is still not clear that the neurological effects of NF-κB reflect transcriptional activation in neurons. Most manipulations and assays are performed in the mixed-cell environments found in vivo, in "neuronal" cell cultures that contain significant numbers of glia, or in tumor-derived "neuronal" cell lines. When transfections or other manipulations have been targeted specifically at neurons, the endpoints measured are typically electrophysiology or other parameters far removed from gene transcription. Careful tests of NF-κB-dependent transcription in highly purified cultures of neurons generally show little to no NF-κB activity.[52][53]
Some of the reports of NF-κB in neurons appear to have been an artifact of antibody nonspecificity.[54] Of course, artifacts of cell culture—e.g., removal of neurons from the influence of glia—could create spurious results as well. But this has been addressed in at least two coculture approaches. Moerman et al.[55] used a coculture format whereby neurons and glia could be separated after treatment for EMSA analysis, and they found that the NF-κB induced by glutamatergic stimuli was restricted to glia (and, intriguingly, only glia that had been in the presence of neurons for 48 hours). The same investigators explored the issue in another approach, utilizing neurons from an NF-κB reporter transgenic mouse cultured with wild-type glia; glutamatergic stimuli again failed to activate in neurons.[56] Some of the DNA-binding activity noted under certain conditions (particularly that reported as constitutive) appears to result from Sp3 and Sp4 binding to a subset of κB enhancer sequences in neurons.[57] This activity is actually inhibited by glutamate and other conditions that elevate intraneuronal calcium. In the final analysis, the role of NF-κB in neurons remains opaque due to the difficulty of measuring transcription in cells that are simultaneously identified for type. Certainly, learning and memory could be influenced by transcriptional changes in astrocytes and other glial elements. And it should be considered that there could be mechanistic effects of NF-κB aside from direct transactivation of genes.
Clinical significance
Cancers
NF-κB is widely used by eukaryotic cells as a regulator of genes that control cell proliferation and cell survival. As such, many different types of human tumors have misregulated NF-κB: that is, NF-κB is constitutively active. Active NF-κB turns on the expression of genes that keep the cell proliferating and protect the cell from conditions that would otherwise cause it to die via apoptosis. In cancer, proteins that control NF-κB signaling are mutated or aberrantly expressed, leading to defective coordination between the malignant cell and the rest of the organism. This is evident both in metastasis, as well as in the inefficient eradication of the tumor by the immune system.[58]
Normal cells can die when removed from the tissue they belong to, or when their genome cannot operate in harmony with tissue function: these events depend on feedback regulation of NF-κB, and fail in cancer.[59]
Defects in NF-κB results in increased susceptibility to apoptosis leading to increased cell death. This is because NF-κB regulates anti-apoptotic genes especially the TRAF1 and TRAF2 and therefore abrogates the activities of the caspase family of enzymes, which are central to most apoptotic processes.[60]
In tumor cells, NF-κB activity is enhanced. For example, in 41% of Nasopharyngeal carcinoma[61], colorectal cancer, prostate cancer and pancreatic tumors either due to mutations in genes encoding the NF-κB transcription factors themselves or in genes that control NF-κB activity (such as IκB genes); in addition, some tumor cells secrete factors that cause NF-κB to become active[62][63]. Blocking NF-κB can cause tumor cells to stop proliferating, to die, or to become more sensitive to the action of anti-tumor agents[64][65]. Thus, NF-κB is the subject of much active research among pharmaceutical companies as a target for anti-cancer therapy.[66]
However, even though convincing experimental data have identified NF-κB as a critical promoter of tumorigenesis, which creates a solid rationale for the development of antitumor therapy that is based upon suppression of NF-κB activity, caution should be exercised when considering anti-NF-κB activity as a broad therapeutic strategy in cancer treatment as data has also shown that NF-κB activity enhances tumor cell sensitivity to apoptosis and senescence. In addition, it has been shown that canonical NF-κB is a Fas transcription activator and the alternative NF-κB is a Fas transcription repressor.[67] Therefore, NF-κB promotes Fas-mediated apoptosis in cancer cells, and thus inhibition of NF-κB may suppress Fas-mediated apoptosis to impair host immune cell-mediated tumor suppression.
Inflammation
Because NF-κB controls many genes involved in inflammation, it is not surprising that NF-κB is found to be chronically active in many inflammatory diseases, such as inflammatory bowel disease, arthritis, sepsis, gastritis, asthma, atherosclerosis[68] and others. It is important to note though, that elevation of some NF-κB activators, such as osteoprotegerin (OPG), are associated with elevated mortality, especially from cardiovascular diseases.[69][70] Elevated NF-κB has also been associated with schizophrenia.[71] Recently, NF-κB activation has been suggested as a possible molecular mechanism for the catabolic effects of cigarette smoke in skeletal muscle and sarcopenia.[72] Research has shown that during inflammation the function of a cell depends on signals it activates in response to contact with adjacent cells and to combinations of hormones, especially cytokines that act on it through specific receptors.[73] A cell’s phenotype within a tissue develops through mutual stimulation of feedback signals that coordinate its function with other cells; this is especially evident during reprogramming of cell function when a tissue is exposed to inflammation, because cells alter their phenotype, and gradually express combinations of genes that prepare the tissue for regeneration after the cause of inflammation is removed.[73][74] Particularly important are feedback responses that develop between tissue resident cells, and circulating cells of the immune system.[74]
Fidelity of feedback responses between diverse cell types and the immune system depends on the integrity of mechanisms that limit the range of genes activated by NF-κB, allowing only expression of genes which contribute to an effective immune response and subsequently, a complete restoration of tissue function after resolution of inflammation.[74] In cancer, mechanisms that regulate gene expression in response to inflammatory stimuli are altered to the point that a cell ceases to link its survival with the mechanisms that coordinate its phenotype and its function with the rest of the tissue.[59] This is often evident in severely compromised regulation of NF-κB activity, which allows cancer cells to express abnormal cohorts of NF-κB target genes.[75] This results in not only the cancer cells functioning abnormally: cells of surrounding tissue alter their function and cease to support the organism exclusively. Additionally, several types of cells in the microenvironment of cancer may change their phenotypes to support cancer growth.[76][77][78] Inflammation, therefore, is a process that tests the fidelity of tissue components because the process that leads to tissue regeneration requires coordination of gene expression between diverse cell types.[73][79]
NEMO
NEMO deficiency syndrome is a rare genetic condition relating to a fault in IKBKG that in turn activates NF-kB. It mostly affects males and has a highly variable set of symptoms and prognoses.[80]
Addiction
NF-κB is one of several induced transcriptional targets of ΔFosB which facilitates the development and maintenance of an addiction to a stimulus.[81][82][83] In the caudate putamen, NF-κB induction is associated with increases in locomotion, whereas in the nucleus accumbens, NF-κB induction enhances the positive reinforcing effect of a drug through reward sensitization.[82]
| Target gene |
Target expression |
Neural effects | Behavioral effects |
|---|---|---|---|
| c-Fos | ↓ | Molecular switch enabling the chronic induction of ΔFosB[note 1] | – |
| dynorphin | ↓ [note 2] | • Downregulation of κ-opioid feedback loop | • Decreased drug aversion |
| NF-κB | ↑ | • Expansion of NAcc dendritic processes • NF-κB inflammatory response in the NAcc • NF-κB inflammatory response in the CP | • Increased drug reward • Increased drug reward • Locomotor sensitization |
| GluR2 | ↑ | • Decreased sensitivity to glutamate | • Increased drug reward |
| Cdk5 | ↑ | • GluR1 synaptic protein phosphorylation • Expansion of NAcc dendritic processes | Decreased drug reward (net effect) |
Non-drug inhibitors
Many natural products (including anti-oxidants) that have been promoted to have anti-cancer and anti-inflammatory activity have also been shown to inhibit NF-κB. There is a controversial US patent (US patent 6,410,516)[85] that applies to the discovery and use of agents that can block NF-κB for therapeutic purposes. This patent is involved in several lawsuits, including Ariad v. Lilly. Recent work by Karin,[86] Ben-Neriah[87] and others has highlighted the importance of the connection between NF-κB, inflammation, and cancer, and underscored the value of therapies that regulate the activity of NF-κB.[88]
Extracts from a number of herbs and dietary plants are efficient inhibitors of NF-κB activation in vitro.[89] Nobiletin, a flavonoid isolated from citrus peels, has been shown to inhibit the NF-κB signaling pathway in mice.[90] The circumsporozoite protein of Plasmodium falciparum has been shown to be an inhibitor of NF-κB.[91]
As a drug target
Aberrant activation of NF-κB is frequently observed in many cancers. Moreover, suppression of NF-κB limits the proliferation of cancer cells. In addition, NF-κB is a key player in the inflammatory response. Hence methods of inhibiting NF-κB signaling has potential therapeutic application in cancer and inflammatory diseases.[92][93]
Both the canonical and non-canonical NF-κB pathways require proteasomal degradation of regulatory pathway components for NF-κB signalling to occur. The proteosome inhibitor Bortezomib broadly blocks this activity and is approved for treatment of NF-κB driven Mantle Cell Lymphoma and Multiple Myeloma[94][95].
The discovery that activation of NF-κB nuclear translocation can be separated from the elevation of oxidant stress[96] gives a promising avenue of development for strategies targeting NF-κB inhibition.
The drug denosumab acts to raise bone mineral density and reduce fracture rates in many patient sub-groups by inhibiting RANKL. RANKL acts through its receptor RANK, which in turn promotes NF-κB,[97] RANKL normally works by enabling the differentiation of osteoclasts from monocytes.
Disulfiram, olmesartan and dithiocarbamates can inhibit the nuclear factor-κB (NF-κB) signaling cascade.[98] Effort to develop direct NF-kB inhibitor has emerged with compounds such as (-)-DHMEQ, PBS-1086, IT-603 and IT-901.[99][100][101] (-)-DHMEQ and PBS-1086 are irreversible binder to NF-KB while IT-603 and IT-901 are reversible binder. DHMEQ covalently binds to Cys 38 of p65.[102]
Anatabine's antiinflammatory effects are claimed to result from modulation of NF-κB activity.[103] However the studies purporting its benefit use abnormally high doses in the millimolar range (similar to the extracellular potassium concentration), which are unlikely to be achieved in humans.
BAY 11-7082 has also been identified as a drug that can inhibit the NF-kB signaling cascade. It is capable of preventing the phosphorylation of IKK-α in an irreversible manner such that there is down regulation of NF-kB activation.[104] In has been shown that administration of BAY 11-7082 rescued renal functionality in diabetic-induced Sprague-Dawley rats by suppressing NF-kB regulated oxidative stress.[105]
In has been shown that administration of BAY 11-7082 rescued renal functionality in diabetic-induced Sprague-Dawley rats by suppressing NF-kB regulated oxidative stress.[106]
Research has shown that the N-acylethanolamine, palmitoylethanolamide is capable of PPAR-mediated inhibition of NF-κB. [107]
The biological target of iguratimod, a drug marketed to treat rheumatoid arthritis in Japan and China, was unknown as of 2015, but the primary mechanism of action appeared to be preventing NF-κB activation.[108]
See also
Notes
- In other words, c-Fos repression allows ΔFosB to accumulate within nucleus accumbens medium spiny neurons more rapidly because it is selectively induced in this state.[83]
- ΔFosB has been implicated in causing both increases and decreases in dynorphin expression in different studies;[82][84] this table entry reflects only a decrease.
References
- Gilmore TD (October 2006). "Introduction to NF-κB: players, pathways, perspectives". Oncogene. 25 (51): 6680–4. doi:10.1038/sj.onc.1209954. PMID 17072321.
- Brasier AR (2006). "The NF-κB regulatory network". Cardiovascular Toxicology. 6 (2): 111–30. doi:10.1385/CT:6:2:111. PMID 17303919.
- Perkins ND (January 2007). "Integrating cell-signalling pathways with NF-κB and IKK function". Nature Reviews Molecular Cell Biology. 8 (1): 49–62. doi:10.1038/nrm2083. PMID 17183360.
- Concetti J, Wilson CL (September 2018). "NFKB1 and Cancer: Friend or Foe?". Cells. 7 (9). doi:10.3390/cells7090133. PMC 6162711. PMID 30205516.
- Gilmore TD (November 1999). "The Rel/NF-κB signal transduction pathway: introduction". Oncogene. 18 (49): 6842–4. doi:10.1038/sj.onc.1203237. PMID 10602459.
- Tian B, Brasier AR (2003). "Identification of a nuclear factor κB-dependent gene network". Recent Progress in Hormone Research. 58: 95–130. doi:10.1210/rp.58.1.95. PMID 12795416.
- Albensi BC, Mattson MP (February 2000). "Evidence for the involvement of TNF and NF-κB in hippocampal synaptic plasticity". Synapse. 35 (2): 151–9. doi:10.1002/(SICI)1098-2396(200002)35:2<151::AID-SYN8>3.0.CO;2-P. PMID 10611641.
- Meffert MK, Chang JM, Wiltgen BJ, Fanselow MS, Baltimore D (October 2003). "NF-κB functions in synaptic signaling and behavior" (PDF). Nature Neuroscience. 6 (10): 1072–8. doi:10.1038/nn1110. PMID 12947408.
- Levenson JM, Choi S, Lee SY, Cao YA, Ahn HJ, Worley KC, Pizzi M, Liou HC, Sweatt JD (April 2004). "A bioinformatics analysis of memory consolidation reveals involvement of the transcription factor c-rel". The Journal of Neuroscience. 24 (16): 3933–43. doi:10.1523/JNEUROSCI.5646-03.2004. PMC 6729420. PMID 15102909.
- Freudenthal R, Locatelli F, Hermitte G, Maldonado H, Lafourcade C, Delorenzi A, Romano A (February 1998). "κ-B like DNA-binding activity is enhanced after spaced training that induces long-term memory in the crab Chasmagnathus". Neuroscience Letters. 242 (3): 143–6. doi:10.1016/S0304-3940(98)00059-7. PMID 9530926.
- Merlo E, Freudenthal R, Romano A (2002). "The IκB kinase inhibitor sulfasalazine impairs long-term memory in the crab Chasmagnathus". Neuroscience. 112 (1): 161–72. doi:10.1016/S0306-4522(02)00049-0. PMID 12044481.
- Park HJ, Youn HS (March 2013). "Mercury induces the expression of cyclooxygenase-2 and inducible nitric oxide synthase". Toxicology and Industrial Health. 29 (2): 169–74. doi:10.1177/0748233711427048. PMID 22080037.
- Sen R, Baltimore D (August 1986). "Multiple nuclear factors interact with the immunoglobulin enhancer sequences". Cell. 46 (5): 705–16. doi:10.1016/0092-8674(86)90346-6. PMID 3091258.
- Karin M, Ben-Neriah Y (2000). "Phosphorylation meets ubiquitination: the control of NF-κB activity". Annual Review of Immunology. 18: 621–63. doi:10.1146/annurev.immunol.18.1.621. PMID 10837071.
- Senftleben U, Cao Y, Xiao G, Greten FR, Krähn G, Bonizzi G, Chen Y, Hu Y, Fong A, Sun SC, Karin M (August 2001). "Activation by IKKα of a second, evolutionary conserved, NF-κB signaling pathway". Science. 293 (5534): 1495–9. doi:10.1126/science.1062677. PMID 11520989.
- Plaksin D, Baeuerle PA, Eisenbach L (June 1993). "KBF1 (p50 NF-κB homodimer) acts as a repressor of H-2Kb gene expression in metastatic tumor cells". The Journal of Experimental Medicine. 177 (6): 1651–62. doi:10.1084/jem.177.6.1651. PMC 2191052. PMID 8496683.
- Guan H, Hou S, Ricciardi RP (March 2005). "DNA binding of repressor nuclear factor-κB p50/p50 depends on phosphorylation of Ser337 by the protein kinase A catalytic subunit". The Journal of Biological Chemistry. 280 (11): 9957–62. doi:10.1074/jbc.m412180200. PMID 15642694.
- Nabel GJ, Verma IM (November 1993). "Proposed NF-κB/IκB family nomenclature". Genes & Development. 7 (11): 2063. doi:10.1101/gad.7.11.2063. PMID 8224837.
- Ghosh S, May MJ, Kopp EB (1998). "NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses". Annual Review of Immunology. 16: 225–60. doi:10.1146/annurev.immunol.16.1.225. PMID 9597130.
- Waterhouse RM, Kriventseva EV, Meister S, Xi Z, Alvarez KS, Bartholomay LC, Barillas-Mury C, Bian G, Blandin S, Christensen BM, Dong Y, Jiang H, Kanost MR, Koutsos AC, Levashina EA, Li J, Ligoxygakis P, Maccallum RM, Mayhew GF, Mendes A, Michel K, Osta MA, Paskewitz S, Shin SW, Vlachou D, Wang L, Wei W, Zheng L, Zou Z, Severson DW, Raikhel AS, Kafatos FC, Dimopoulos G, Zdobnov EM, Christophides GK (June 2007). "Evolutionary dynamics of immune-related genes and pathways in disease-vector mosquitoes". Science. 316 (5832): 1738–43. Bibcode:2007Sci...316.1738W. doi:10.1126/science.1139862. PMC 2042107. PMID 17588928.
- PDB: 3do7; Fusco AJ, Huang DB, Miller D, Wang VY, Vu D, Ghosh G (February 2009). "NF-κB p52:RelB heterodimer recognizes two classes of κB sites with two distinct modes". EMBO Reports. 10 (2): 152–9. doi:10.1038/embor.2008.227. PMC 2637311. PMID 19098713.
- (a) Chandel NS, Trzyna WC, McClintock DS, Schumacker PT (July 2000). "Role of oxidants in NF-κB activation and TNF-α gene transcription induced by hypoxia and endotoxin". Journal of Immunology. 165 (2): 1013–21. doi:10.4049/jimmunol.165.2.1013. PMID 10878378.; (b) Fitzgerald DC, Meade KG, McEvoy AN, Lillis L, Murphy EP, MacHugh DE, Baird AW (March 2007). "Tumour necrosis factor-α (TNF-α) increases nuclear factor κB (NFκB) activity in and interleukin-8 (IL-8) release from bovine mammary epithelial cells". Veterinary Immunology and Immunopathology. 116 (1–2): 59–68. doi:10.1016/j.vetimm.2006.12.008. PMID 17276517.; (c) Renard P, Zachary MD, Bougelet C, Mirault ME, Haegeman G, Remacle J, Raes M (January 1997). "Effects of antioxidant enzyme modulations on interleukin-1-induced nuclear factor κB activation". Biochemical Pharmacology. 53 (2): 149–60. doi:10.1016/S0006-2952(96)00645-4. PMID 9037247.; (d) Qin H, Wilson CA, Lee SJ, Zhao X, Benveniste EN (November 2005). "LPS induces CD40 gene expression through the activation of NF-κB and STAT-1α in macrophages and microglia". Blood. 106 (9): 3114–22. doi:10.1182/blood-2005-02-0759. PMC 1895321. PMID 16020513.; (e) Takemoto Y, Yoshiyama M, Takeuchi K, Omura T, Komatsu R, Izumi Y, Kim S, Yoshikawa J (November 1999). "Increased JNK, AP-1 and NF-κB DNA binding activities in isoproterenol-induced cardiac remodeling". Journal of Molecular and Cellular Cardiology. 31 (11): 2017–30. doi:10.1006/jmcc.1999.1033. PMID 10591028.; (f) Hargrave BY, Tiangco DA, Lattanzio FA, Beebe SJ (2003). "Cocaine, not morphine, causes the generation of reactive oxygen species and activation of NF-κB in transiently cotransfected heart cells". Cardiovascular Toxicology. 3 (2): 141–51. doi:10.1385/CT:3:2:141. PMID 14501032.; (g) Basu S, Rosenzweig KR, Youmell M, Price BD (June 1998). "The DNA-dependent protein kinase participates in the activation of NF κB following DNA damage". Biochemical and Biophysical Research Communications. 247 (1): 79–83. doi:10.1006/bbrc.1998.8741. PMID 9636658.
- Baud'huin M, Lamoureux F, Duplomb L, Rédini F, Heymann D (September 2007). "RANKL, RANK, osteoprotegerin: key partners of osteoimmunology and vascular diseases". Cellular and Molecular Life Sciences. 64 (18): 2334–50. doi:10.1007/s00018-007-7104-0. PMID 17530461.
- Doyle SL, O'Neill LA (October 2006). "Toll-like receptors: from the discovery of NFκB to new insights into transcriptional regulations in innate immunity". Biochemical Pharmacology. 72 (9): 1102–13. doi:10.1016/j.bcp.2006.07.010. PMID 16930560.
- Hayden MS, West AP, Ghosh S (October 2006). "NF-κB and the immune response". Oncogene. 25 (51): 6758–80. doi:10.1038/sj.onc.1209943. PMID 17072327.
- Li Q, Verma IM (October 2002). "NF-κB regulation in the immune system". Nature Reviews. Immunology. 2 (10): 725–34. doi:10.1038/nri910. PMID 12360211.
- Fujita T, Nolan GP, Liou HC, Scott ML, Baltimore D (July 1993). "The candidate proto-oncogene bcl-3 encodes a transcriptional coactivator that activates through NF-κB p50 homodimers". Genes & Development. 7 (7B): 1354–63. doi:10.1101/gad.7.7b.1354. PMID 8330739.
- Franzoso G, Bours V, Park S, Tomita-Yamaguchi M, Kelly K, Siebenlist U (September 1992). "The candidate oncoprotein Bcl-3 is an antagonist of p50/NF-κB-mediated inhibition". Nature. 359 (6393): 339–42. doi:10.1038/359339a0. PMID 1406939.
- Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, Siebenlist U (March 1993). "The oncoprotein Bcl-3 directly transactivates through κB motifs via association with DNA-binding p50B homodimers". Cell. 72 (5): 729–39. doi:10.1016/0092-8674(93)90401-B. PMID 8453667.
- Jacobs MD, Harrison SC (December 1998). "Structure of an IκBα/NF-κB complex". Cell. 95 (6): 749–58. doi:10.1016/S0092-8674(00)81698-0. PMID 9865693.
- Basak S, Kim H, Kearns JD, Tergaonkar V, O'Dea E, Werner SL, Benedict CA, Ware CF, Ghosh G, Verma IM, Hoffmann A (January 2007). "A fourth IκB protein within the NF-κB signaling module". Cell. 128 (2): 369–81. doi:10.1016/j.cell.2006.12.033. PMC 1831796. PMID 17254973..
- Dobrzanski P, Ryseck RP, Bravo R (March 1995). "Specific inhibition of RelB/p52 transcriptional activity by the C-terminal domain of p100". Oncogene. 10 (5): 1003–7. PMID 7898917.
- Lo JC, Basak S, James ES, Quiambo RS, Kinsella MC, Alegre ML, Weih F, Franzoso G, Hoffmann A, Fu YX (February 2006). "Coordination between NF-κB family members p50 and p52 is essential for mediating LTβR signals in the development and organization of secondary lymphoid tissues". Blood. 107 (3): 1048–55. doi:10.1182/blood-2005-06-2452. PMC 1895903. PMID 16195333.
- Deptala A, Bedner E, Gorczyca W, Darzynkiewicz Z (November 1998). "Activation of nuclear factor κB (NF-κB) assayed by laser scanning cytometry (LSC)". Cytometry. 33 (3): 376–82. doi:10.1002/(SICI)1097-0320(19981101)33:3<376::AID-CYTO13>3.0.CO;2-Q. PMC 3874872. PMID 9822350.
- Nelson DE, Ihekwaba AE, Elliott M, Johnson JR, Gibney CA, Foreman BE, Nelson G, See V, Horton CA, Spiller DG, Edwards SW, McDowell HP, Unitt JF, Sullivan E, Grimley R, Benson N, Broomhead D, Kell DB, White MR (October 2004). "Oscillations in NF-κB signaling control the dynamics of gene expression". Science. 306 (5696): 704–8. doi:10.1126/science.1099962. PMID 15499023.
- Hiscott J, Kwon H, Génin P (January 2001). "Hostile takeovers: viral appropriation of the NF-κB pathway". The Journal of Clinical Investigation. 107 (2): 143–51. doi:10.1172/JCI11918. PMC 199181. PMID 11160127.
- Adkins I, Schulz S, Borgmann S, Autenrieth IB, Gröbner S (February 2008). "Differential roles of Yersinia outer protein P-mediated inhibition of nuclear factor-κB in the induction of cell death in dendritic cells and macrophages". Journal of Medical Microbiology. 57 (Pt 2): 139–44. doi:10.1099/jmm.0.47437-0. PMID 18201977.
- Micheli L, Leonardi L, Conti F, Buanne P, Canu N, Caruso M, Tirone F (March 2005). "PC4 coactivates MyoD by relieving the histone deacetylase 4-mediated inhibition of myocyte enhancer factor 2C". Molecular and Cellular Biology. 25 (6): 2242–59. doi:10.1128/MCB.25.6.2242-2259.2005. PMC 1061592. PMID 15743821.
- Micheli L, Leonardi L, Conti F, Maresca G, Colazingari S, Mattei E, Lira SA, Farioli-Vecchioli S, Caruso M, Tirone F (February 2011). "PC4/Tis7/IFRD1 stimulates skeletal muscle regeneration and is involved in myoblast differentiation as a regulator of MyoD and NF-κB". The Journal of Biological Chemistry. 286 (7): 5691–707. doi:10.1074/jbc.M110.162842. PMC 3037682. PMID 21127072.
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW (June 2004). "Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase". The EMBO Journal. 23 (12): 2369–80. doi:10.1038/sj.emboj.7600244. PMC 423286. PMID 15152190.
- Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, Karin M (October 2004). "Activation of IKKα target genes depends on recognition of specific κB binding sites by RelB:p52 dimers". The EMBO Journal. 23 (21): 4202–10. doi:10.1038/sj.emboj.7600391. PMC 524385. PMID 15470505.
- Basak S, Shih VF, Hoffmann A (May 2008). "Generation and activation of multiple dimeric transcription factors within the NF-κB signaling system". Molecular and Cellular Biology. 28 (10): 3139–50. doi:10.1128/MCB.01469-07. PMC 2423155. PMID 18299388.
- Smith EM, Gregg M, Hashemi F, Schott L, Hughes TK (2006-07-01). "Corticotropin Releasing Factor (CRF) activation of NF-κB-directed transcription in leukocytes". Cellular and Molecular Neurobiology. 26 (4–6): 1021–36. doi:10.1007/s10571-006-9040-1. PMID 16633893.
- Livolsi A, Busuttil V, Imbert V, Abraham RT, Peyron JF (March 2001). "Tyrosine phosphorylation-dependent activation of NF-κB. Requirement for p56 LCK and ZAP-70 protein tyrosine kinases". European Journal of Biochemistry. 268 (5): 1508–15. doi:10.1046/j.1432-1327.2001.02028.x. PMID 11231305.
- Heckscher ES, Fetter RD, Marek KW, Albin SD, Davis GW (September 2007). "NF-κB, IκB, and IRAK control glutamate receptor density at the Drosophila NMJ". Neuron. 55 (6): 859–73. doi:10.1016/j.neuron.2007.08.005. PMC 2701504. PMID 17880891.
- Kaltschmidt B, Ndiaye D, Korte M, Pothion S, Arbibe L, Prüllage M, Pfeiffer J, Lindecke A, Staiger V, Israël A, Kaltschmidt C, Mémet S (April 2006). "NF-κB regulates spatial memory formation and synaptic plasticity through protein kinase A/CREB signaling". Molecular and Cellular Biology. 26 (8): 2936–46. doi:10.1128/MCB.26.8.2936-2946.2006. PMC 1446931. PMID 16581769.
- Wang J, Fu XQ, Lei WL, Wang T, Sheng AL, Luo ZG (August 2010). "Nuclear factor κB controls acetylcholine receptor clustering at the neuromuscular junction". The Journal of Neuroscience. 30 (33): 11104–13. doi:10.1523/JNEUROSCI.2118-10.2010. PMC 6633475. PMID 20720118.
- Boersma MC, Dresselhaus EC, De Biase LM, Mihalas AB, Bergles DE, Meffert MK (April 2011). "A requirement for nuclear factor-κB in developmental and plasticity-associated synaptogenesis". The Journal of Neuroscience. 31 (14): 5414–25. doi:10.1523/JNEUROSCI.2456-10.2011. PMC 3113725. PMID 21471377.
- Gutierrez H, Hale VA, Dolcet X, Davies A (April 2005). "NF-κB signalling regulates the growth of neural processes in the developing PNS and CNS". Development. 132 (7): 1713–26. doi:10.1242/dev.01702. PMID 15743881.
- Zaheer A, Yorek MA, Lim R (December 2001). "Effects of glia maturation factor overexpression in primary astrocytes on MAP kinase activation, transcription factor activation, and neurotrophin secretion". Neurochemical Research. 26 (12): 1293–9. doi:10.1023/A:1014241300179. PMID 11885780.
- Qiu J, Hu X, Nesic O, Grafe MR, Rassin DK, Wood TG, Perez-Polo JR (July 2004). "Effects of NF-κB oligonucleotide "decoys" on gene expression in P7 rat hippocampus after hypoxia/ischemia". Journal of Neuroscience Research. 77 (1): 108–18. doi:10.1002/jnr.20156. PMID 15197744.
- Listwak SJ, Rathore P, Herkenham M (October 2013). "Minimal NF-κB activity in neurons". Neuroscience. 250: 282–99. doi:10.1016/j.neuroscience.2013.07.013. PMC 3785079. PMID 23872390.
- Jarosinski KW, Whitney LW, Massa PT (September 2001). "Specific deficiency in nuclear factor-κB activation in neurons of the central nervous system". Laboratory Investigation; A Journal of Technical Methods and Pathology. 81 (9): 1275–88. doi:10.1038/labinvest.3780341. PMID 11555675.
- Herkenham M, Rathore P, Brown P, Listwak SJ (October 2011). "Cautionary notes on the use of NF-κB p65 and p50 antibodies for CNS studies". Journal of Neuroinflammation. 8: 141. doi:10.1186/1742-2094-8-141. PMC 3210105. PMID 21999414.
- Moerman AM, Mao X, Lucas MM, Barger SW (April 1999). "Characterization of a neuronal κB-binding factor distinct from NF-κB". Brain Research. Molecular Brain Research. 67 (2): 303–15. doi:10.1016/s0169-328x(99)00091-1. PMID 10216229.
- Mao XR, Moerman-Herzog AM, Chen Y, Barger SW (May 2009). "Unique aspects of transcriptional regulation in neurons--nuances in NFκB and Sp1-related factors". Journal of Neuroinflammation. 6: 16. doi:10.1186/1742-2094-6-16. PMC 2693111. PMID 19450264.
- Mao X, Yang SH, Simpkins JW, Barger SW (March 2007). "Glutamate receptor activation evokes calpain-mediated degradation of Sp3 and Sp4, the prominent Sp-family transcription factors in neurons". Journal of Neurochemistry. 100 (5): 1300–14. doi:10.1111/j.1471-4159.2006.04297.x. PMC 1949346. PMID 17316402.
- Vlahopoulos, SA (August 2017). "Aberrant control of NF-κB in cancer permits transcriptional and phenotypic plasticity, to curtail dependence on host tissue: molecular mode". Cancer Biology & Medicine. 14 (3): 254–270. doi:10.20892/j.issn.2095-3941.2017.0029. PMC 5570602. PMID 28884042.
- Vlahopoulos SA, Cen O, Hengen N, Agan J, Moschovi M, Critselis E, Adamaki M, Bacopoulou F, Copland JA, Boldogh I, Karin M, Chrousos GP (August 2015). "Dynamic aberrant NF-κB spurs tumorigenesis: a new model encompassing the microenvironment". Cytokine & Growth Factor Reviews. 26 (4): 389–403. doi:10.1016/j.cytogfr.2015.06.001. PMC 4526340. PMID 26119834.
- Sheikh MS, Huang Y (2003). "Death receptor activation complexes: it takes two to activate TNF receptor 1". Cell Cycle. 2 (6): 550–2. doi:10.4161/cc.2.6.566. PMID 14504472.
- Li YY, Chung GT, Lui VW, To KF, Ma BB, Chow C, et al. (January 2017). "Exome and genome sequencing of nasopharynx cancer identifies NF-κB pathway activating mutations". Nature Communications. 8: 14121. Bibcode:2017NatCo...814121L. doi:10.1038/ncomms14121. PMC 5253631. PMID 28098136.
- Sun SC (January 2011). "Non-canonical NF-κB signaling pathway". Cell Research. 21 (1): 71–85. doi:10.1038/cr.2010.177. PMC 3193406. PMID 21173796.
- Nouri M, Massah S, Caradec J, Lubik AA, Li N, Truong S, et al. (April 2020). "Transient Sox9 Expression Facilitates Resistance to Androgen-Targeted Therapy in Prostate Cancer". Clinical Cancer Research. 26 (7): 1678–1689. doi:10.1158/1078-0432.CCR-19-0098. PMID 31919137.
- Taniguchi K, Karin M (May 2018). "NF-κB, inflammation, immunity and cancer: coming of age". Nature Reviews. Immunology. 18 (5): 309–324. doi:10.1038/nri.2017.142. PMID 29379212.
- Sun L, Mathews LA, Cabarcas SM, Zhang X, Yang A, Zhang Y, et al. (August 2013). "Epigenetic regulation of SOX9 by the NF-κB signaling pathway in pancreatic cancer stem cells". Stem Cells. 31 (8): 1454–66. doi:10.1002/stem.1394. PMC 3775871. PMID 23592398.
- Escárcega RO, Fuentes-Alexandro S, García-Carrasco M, Gatica A, Zamora A (March 2007). "The transcription factor nuclear factor-kappa B and cancer". Clinical Oncology. 19 (2): 154–61. doi:10.1016/j.clon.2006.11.013. PMID 17355113.
- Liu F, Bardhan K, Yang D, Thangaraju M, Ganapathy V, Waller JL, Liles GB, Lee JR, Liu K (July 2012). "NF-κB directly regulates Fas transcription to modulate Fas-mediated apoptosis and tumor suppression". The Journal of Biological Chemistry. 287 (30): 25530–40. doi:10.1074/jbc.M112.356279. PMC 3408167. PMID 22669972.
- Monaco C, Andreakos E, Kiriakidis S, Mauri C, Bicknell C, Foxwell B, Cheshire N, Paleolog E, Feldmann M (April 2004). "Canonical pathway of nuclear factor κB activation selectively regulates proinflammatory and prothrombotic responses in human atherosclerosis". Proceedings of the National Academy of Sciences of the United States of America. 101 (15): 5634–9. doi:10.1073/pnas.0401060101. PMC 397455. PMID 15064395.
- Venuraju SM, Yerramasu A, Corder R, Lahiri A (May 2010). "Osteoprotegerin as a predictor of coronary artery disease and cardiovascular mortality and morbidity". Journal of the American College of Cardiology. 55 (19): 2049–61. doi:10.1016/j.jacc.2010.03.013. PMID 20447527.
- Lieb W, Gona P, Larson MG, Massaro JM, Lipinska I, Keaney JF, Rong J, Corey D, Hoffmann U, Fox CS, Vasan RS, Benjamin EJ, O'Donnell CJ, Kathiresan S (September 2010). "Biomarkers of the osteoprotegerin pathway: clinical correlates, subclinical disease, incident cardiovascular disease, and mortality". Arteriosclerosis, Thrombosis, and Vascular Biology. 30 (9): 1849–54. doi:10.1161/ATVBAHA.109.199661. PMC 3039214. PMID 20448212.
- Song XQ, Lv LX, Li WQ, Hao YH, Zhao JP (March 2009). "The interaction of nuclear factor-κB and cytokines is associated with schizophrenia". Biological Psychiatry. 65 (6): 481–8. doi:10.1016/j.biopsych.2008.10.018. PMID 19058794.
- Kaisari S, Rom O, Aizenbud D, Reznick AZ (2013). "Involvement of NF-κB and muscle specific E3 ubiquitin ligase MuRF1 in cigarette smoke-induced catabolism in C2 myotubes". Advances in Experimental Medicine and Biology. 788: 7–17. doi:10.1007/978-94-007-6627-3_2. ISBN 978-94-007-6626-6. PMID 23835952.
- Hajishengallis G, Chavakis T (January 2013). "Endogenous modulators of inflammatory cell recruitment". Trends in Immunology. 34 (1): 1–6. doi:10.1016/j.it.2012.08.003. PMC 3703146. PMID 22951309.
- Vidal PM, Lemmens E, Dooley D, Hendrix S (February 2013). "The role of "anti-inflammatory" cytokines in axon regeneration". Cytokine & Growth Factor Reviews. 24 (1): 1–12. doi:10.1016/j.cytogfr.2012.08.008. PMID 22985997.
- Grivennikov SI, Karin M (February 2010). "Dangerous liaisons: STAT3 and NF-κB collaboration and crosstalk in cancer". Cytokine & Growth Factor Reviews. 21 (1): 11–9. doi:10.1016/j.cytogfr.2009.11.005. PMC 2834864. PMID 20018552.
- Bonavita E, Galdiero MR, Jaillon S, Mantovani A (2015). "Phagocytes as Corrupted Policemen in Cancer-Related Inflammation". Advances in Cancer Research. 128: 141–71. doi:10.1016/bs.acr.2015.04.013. ISBN 9780128023167. PMID 26216632.
- Sionov RV, Fridlender ZG, Granot Z (December 2015). "The Multifaceted Roles Neutrophils Play in the Tumor Microenvironment". Cancer Microenvironment. 8 (3): 125–58. doi:10.1007/s12307-014-0147-5. PMC 4714999. PMID 24895166.
- Kong X, Li L, Li Z, Xie K (December 2012). "Targeted destruction of the orchestration of the pancreatic stroma and tumor cells in pancreatic cancer cases: molecular basis for therapeutic implications". Cytokine & Growth Factor Reviews. 23 (6): 343–56. doi:10.1016/j.cytogfr.2012.06.006. PMC 3505269. PMID 22749856.
- Mecollari V, Nieuwenhuis B, Verhaagen J (2014). "A perspective on the role of class III semaphorin signaling in central nervous system trauma". Frontiers in Cellular Neuroscience. 8: 328. doi:10.3389/fncel.2014.00328. PMC 4209881. PMID 25386118.
- NEMO deficiency syndrome information, Great Ormond Street Hospital for Children
- Robison AJ, Nestler EJ (October 2011). "Transcriptional and epigenetic mechanisms of addiction". Nature Reviews. Neuroscience. 12 (11): 623–37. doi:10.1038/nrn3111. PMC 3272277. PMID 21989194.
- Ruffle JK (November 2014). "Molecular neurobiology of addiction: what's all the (Δ)FosB about?". The American Journal of Drug and Alcohol Abuse. 40 (6): 428–37. doi:10.3109/00952990.2014.933840. PMID 25083822.
- Nestler EJ (December 2013). "Cellular basis of memory for addiction". Dialogues in Clinical Neuroscience. 15 (4): 431–43. PMC 3898681. PMID 24459410.
- Nestler EJ (October 2008). "Review. Transcriptional mechanisms of addiction: role of ΔFosB". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 363 (1507): 3245–55. doi:10.1098/rstb.2008.0067. PMC 2607320. PMID 18640924.
Table 3 - US patent 6410516, Baltimore D; Sen R; Sharp PA; Singh H; Staudt L; Lebowitz JH; Baldwin Jr AS; Clerc RG; Corcoran LM; Baeuerle PA; Lenardo MJ; Fan C-M; Maniatis TPD, "Nuclear factors associated with transcriptional regulation", issued 2002-06-25
- Karin M (March 2008). "The IκB kinase - a bridge between inflammation and cancer". Cell Research. 18 (3): 334–42. doi:10.1038/cr.2008.30. PMID 18301380.
- Pikarsky E, Ben-Neriah Y (April 2006). "NF-κB inhibition: a double-edged sword in cancer?". European Journal of Cancer. 42 (6): 779–84. doi:10.1016/j.ejca.2006.01.011. PMID 16530406.
- Mantovani A, Marchesi F, Portal C, Allavena P, Sica A (2008). "Linking inflammation reactions to cancer: novel targets for therapeutic strategies". Advances in Experimental Medicine and Biology. 610: 112–27. doi:10.1007/978-0-387-73898-7_9. ISBN 978-0-387-73897-0. PMID 18593019.
- Paur I, Balstad TR, Kolberg M, Pedersen MK, Austenaa LM, Jacobs DR, Blomhoff R (May 2010). "Extract of oregano, coffee, thyme, clove, and walnuts inhibits NF-κB in monocytes and in transgenic reporter mice". Cancer Prevention Research. 3 (5): 653–63. doi:10.1158/1940-6207.CAPR-09-0089. PMID 20424131.
- Lin Z, Wu D, Huang L, Jiang C, Pan T, Kang X, Pan J (2019). "via Suppression of NF-κB Signaling and Attenuates Osteoarthritis in Mice". Frontiers in Pharmacology. 10: 570. doi:10.3389/fphar.2019.00570. PMC 6554687. PMID 31214026.
- Ding Y, Huang X, Liu T, Fu Y, Tan Z, Zheng H, Zhou T, Dai J, Xu W (October 2012). "The Plasmodium circumsporozoite protein, a novel NF-κB inhibitor, suppresses the growth of SW480". Pathology Oncology Research. 18 (4): 895–902. doi:10.1007/s12253-012-9519-7. PMID 22678765.
- Garg A, Aggarwal BB (June 2002). "Nuclear transcription factor-κB as a target for cancer drug development". Leukemia. 16 (6): 1053–68. doi:10.1038/sj.leu.2402482. PMID 12040437.
- Sethi G, Sung B, Aggarwal BB (January 2008). "Nuclear factor-κB activation: from bench to bedside". Experimental Biology and Medicine. 233 (1): 21–31. doi:10.3181/0707-MR-196. PMID 18156302.
- Curran MP, McKeage K (2009). "Bortezomib: a review of its use in patients with multiple myeloma". Drugs. 69 (7): 859–88. doi:10.2165/00003495-200969070-00006. PMID 19441872.
- Raedler L (March 2015). "Velcade (Bortezomib) Receives 2 New FDA Indications: For Retreatment of Patients with Multiple Myeloma and for First-Line Treatment of Patients with Mantle-Cell Lymphoma". American Health & Drug Benefits. 8 (Spec Feature): 135–40. PMC 4665054. PMID 26629279.
- Vlahopoulos S, Boldogh I, Casola A, Brasier AR (September 1999). "Nuclear factor-κB-dependent induction of interleukin-8 gene expression by tumor necrosis factor alpha: evidence for an antioxidant sensitive activating pathway distinct from nuclear translocation". Blood. 94 (6): 1878–89. doi:10.1182/blood.V94.6.1878.418k03_1878_1889. PMID 10477716.
- Hamdy NA (January 2008). "Denosumab: RANKL inhibition in the management of bone loss". Drugs of Today. 44 (1): 7–21. doi:10.1358/dot.2008.44.1.1178467. PMID 18301800.
- Cvek B, Dvorak Z (2007). "Targeting of nuclear factor-κB and proteasome by dithiocarbamate complexes with metals". Current Pharmaceutical Design. 13 (30): 3155–67. doi:10.2174/138161207782110390. PMID 17979756.
- Blakely CM, Pazarentzos E, Olivas V, Asthana S, Yan JJ, Tan I, Hrustanovic G, Chan E, Lin L, Neel DS, Newton W, Bobb KL, Fouts TR, Meshulam J, Gubens MA, Jablons DM, Johnson JR, Bandyopadhyay S, Krogan NJ, Bivona TG (April 2015). "NF-κB-activating complex engaged in response to EGFR oncogene inhibition drives tumor cell survival and residual disease in lung cancer". Cell Reports. 11 (1): 98–110. doi:10.1016/j.celrep.2015.03.012. PMC 4394036. PMID 25843712.
- Fabre C, Mimura N, Bobb K, Kong SY, Gorgun G, Cirstea D, Hu Y, Minami J, Ohguchi H, Zhang J, Meshulam J, Carrasco RD, Tai YT, Richardson PG, Hideshima T, Anderson KC (September 2012). "Dual inhibition of canonical and noncanonical NF-κB pathways demonstrates significant antitumor activities in multiple myeloma". Clinical Cancer Research. 18 (17): 4669–81. doi:10.1158/1078-0432.CCR-12-0779. PMC 4456190. PMID 22806876.
- Shono Y, Tuckett AZ, Liou HC, Doubrovina E, Derenzini E, Ouk S, Tsai JJ, Smith OM, Levy ER, Kreines FM, Ziegler CG, Scallion MI, Doubrovin M, Heller G, Younes A, O'Reilly RJ, van den Brink MR, Zakrzewski JL (January 2016). "Characterization of a c-Rel Inhibitor That Mediates Anticancer Properties in Hematologic Malignancies by Blocking NF-κB-Controlled Oxidative Stress Responses". Cancer Research. 76 (2): 377–89. doi:10.1158/0008-5472.CAN-14-2814. PMC 4715937. PMID 26744524.
- Yamamoto M, Horie R, Takeiri M, Kozawa I, Umezawa K (September 2008). "Inactivation of NF-κB components by covalent binding of (-)-dehydroxymethylepoxyquinomicin to specific cysteine residues". Journal of Medicinal Chemistry. 51 (18): 5780–8. doi:10.1021/jm8006245. PMID 18729348.
- "Role of RCP006 as an anti-inflammatory agent". Roskamp Institute. Retrieved 2011-09-06.
- Kolati SR, Kasala ER, Bodduluru LN, Mahareddy JR, Uppulapu SK, Gogoi R, Barua CC, Lahkar M (March 2015). "BAY 11-7082 ameliorates diabetic nephropathy by attenuating hyperglycemia-mediated oxidative stress and renal inflammation via NF-κB pathway". Environmental Toxicology and Pharmacology. 39 (2): 690–9. doi:10.1016/j.etap.2015.01.019. PMID 25704036.
- Kumar A, Negi G, Sharma SS (May 2012). "Suppression of NF-κB and NF-κB regulated oxidative stress and neuroinflammation by BAY 11-7082 (IκB phosphorylation inhibitor) in experimental diabetic neuropathy". Biochimie. 94 (5): 1158–65. doi:10.1016/j.biochi.2012.01.023. PMID 22342224.
- Kumar A, Negi G, Sharma SS (May 2012). "Suppression of NF-κB and NF-κB regulated oxidative stress and neuroinflammation by BAY 11-7082 (IκB phosphorylation inhibitor) in experimental diabetic neuropathy". Biochimie. 94 (5): 1158–65. doi:10.1016/j.biochi.2012.01.023. PMID 22342224.
- Dana N, Vaseghi G, and Haghjooy Javanmard S (February 2019). "Crosstalk between Peroxisome Proliferator-Activated Receptors and Toll-Like Receptors: A Systematic Review". Advanced Pharmaceutical Bulletin. 39 (2): 690–9. doi:10.15171/apb.2019.003. PMC 6468223. PMID 31011554.
- Tanaka K, Yamaguchi T, Hara M (May 2015). "Iguratimod for the treatment of rheumatoid arthritis in Japan". Expert Review of Clinical Immunology. 11 (5): 565–73. doi:10.1586/1744666X.2015.1027151. PMID 25797025.
External links
- NF-kappa+B at the US National Library of Medicine Medical Subject Headings (MeSH)
- Sankar Ghosh (2006). Handbook of Transcription Factor NF-κB. Boca Raton: CRC. ISBN 978-0-8493-2794-0.
- Thomas D Gilmore. "The Rel/NF-κB Signal Transduction Pathway". Boston University. Retrieved 2007-12-02.
| Addiction |
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Dependence |
| ||||||||||
| Treatment and management |
| ||||||||||
| See also |
| ||||||||||
| |||||||||||