Kv1.1
Potassium voltage-gated channel subfamily A member 1 also known as Kv1.1 is a shaker related voltage-gated potassium channel that in humans is encoded by the KCNA1 gene.[4][5][6] The Isaacs syndrome is a result of an autoimmune reaction against the Kv1.1 ion channel.[7]
Genomics
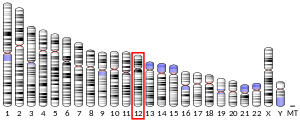
The gene is located on the Watson (plus) strand of the short arm of chromosome 12 (12p13.32). The gene itself is 8,348 bases in length and encodes a protein of 495 amino acids (predicted molecular weight 56.466 kiloDaltons).
Alternative names
The recommended name for this protein is potassium voltage-gated channel subfamily A member 1 but a number of alternatives have been used in the literature including HuK1 (human K+ channel I), RBK1 (rubidium potassium channel 1), MBK (mouse brain K+ channel), voltage gated potassium channel HBK1, voltage gated potassium channel subunit Kv1.1, voltage-gated K+ channel HuKI and AEMK (associated with myokymia with periodic ataxia).
Structure
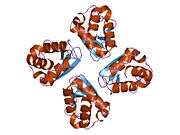
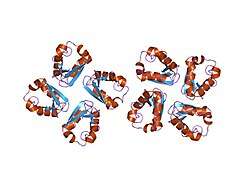
The protein is believed to have six domains (S1-S6) with the loop between S5 and S6 forming the channel pore. This region also has a conserved selectivity filter motif. The functional channel is a homotetramer. The N-terminus of the protein associates with β subunits. These subunits regulate channel inactivation as well as its expression. The C-terminus is associated with a PDZ domain protein involved in channel targeting.[8][9]
Function
The protein functions as a potassium selective channel through which the potassium ion may pass in consensus with the electrochemical gradient. They play a role in repolarisation of membranes.[8]
RNA editing
The pre-mRNA of this protein is subject to RNA editing.[10]
Type
A to I RNA editing is catalyzed by a family of adenosine deaminases acting on RNA (ADARs) that specifically recognize adenosines within double-stranded regions of pre-mRNAs (e.g. Potassium channel RNA editing signal) and deaminate them to inosine. Inosines are recognised as guanosine by the cells translational machinery. There are three members of the ADAR family ADARs 1-3 with ADAR1 and ADAR2 being the only enzymatically active members. ADAR3 is thought to have a regulatory role in the brain. ADAR1 and ADAR2 are widely expressed in tissues while ADAR3 is restricted to the brain. The double stranded regions of RNA are formed by base-pairing between residues in the region close to the editing site with residues usually in a neighboring intron but can sometimes be an exonic sequence too. The region that base pairs with the editing region is known as an Editing Complementary Sequence (ECS).
Location
The modified residue is found at amino acid 400 of the final protein. This is located in the sixth transmembrane region found, which corresponds to the inner vestibule of the pore. A stem loop hairpin structure mediates the RNA editing. ADAR2 is likely to be the preferred editing enzyme at the I/V site. Editing results in a codon alteration from ATT to GTT, resulting in an amino acid change from isoleucine to valine. ADAR2 enzyme is the major editing enzyme. The MFOLD programme predicted that the minimum region required for editing would form an imperfect inverted repeat hairpin. This region is composed of a 114 base pairs. Similar regions have been identified in mouse and rat. The edited adenosine is found in a 6-base pair duplex region. Mutation experiment in the region near the 6-base pair duplex have shown that the specific bases in this region were also essential for editing to occur. The region required for editing is unusual in that the hairpin structure is formed by exonic sequences only. In the majority of A to I editing the ECS is found within an intronic sequence.[10]
Conservation
The editing is highly conserved having been observed in squid, fruit fly, mouse, and rat.[10]
Regulation
Editing levels vary in different tissues: 17% in the caudate nucleus, 68% in the spinal cord, and 77% in the medulla.[11]
Consequences
Structure
Editing results in a codon (I/V) change from (ATT) to (GTT) resulting in translation of a valine instead of an isoleucine at the position of the editing site. Valine has a larger side-chain. RNA editing at this position occurs at a highly conserved ion conducting pore of the channel. This may affect the channels role in the process of fast inactivation.[12]
Function
Voltage-dependent potassium channels modulate excitability by opening and closing a potassium selective pore in response to voltage. The flow of potassium ions is interrupted by interaction of an inactivating particle, an auxiliary protein in humans but an intrinsic part of the channel in other species. The I to V amino acid change is thought to disrupt the hydrophobic interaction between the inactivating particle and the pore lining. This interrupts the process of fast inactivation. Activation kinetics are unaffected by RNA editing.[10] Changes in inactivation kinetics affect the duration and frequency of the action potential. An edited channel passes more current and has a shorter action potential than the non-edited type due to the inability of the inactivating particle to interact with the residue in the ion-conducting pore of the channel. This was determined by electrophysiology analysis.[13] The length of time the membrane is depolarised is decreased, which also reduces the efficiency of transmitter release.[11] Since editing can cause amino acid changes in 1- 4 in potassium channel tetramers, it can have a wide variety of effects on channel inactivation.
Dysregulation
Changes in the process of fast inactivation are known to have behavioral and neurological consequences in vivo.[10]
Clinical
Mutations in this gene cause episodic ataxia type 1.
See also
- GABRA3 - a channel subunit which undergoes similar RNA editing
References
- GRCh38: Ensembl release 89: ENSG00000111262 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Curran ME, Landes GM, Keating MT (1992). "Molecular cloning, characterization, and genomic localization of a human potassium channel gene". Genomics. 12 (4): 729–37. doi:10.1016/0888-7543(92)90302-9. PMID 1349297.
- Albrecht B, Weber K, Pongs O (1995). "Characterization of a voltage-activated K-channel gene cluster on human chromosome 12p13". Recept. Channels. 3 (3): 213–20. PMID 8821794.
- Gutman GA, Chandy KG, Grissmer S, Lazdunski M, McKinnon D, Pardo LA, Robertson GA, Rudy B, Sanguinetti MC, Stühmer W, Wang X (2005). "International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels". Pharmacol. Rev. 57 (4): 473–508. doi:10.1124/pr.57.4.10. PMID 16382104.
- Newsom-Davis J (1997). "Autoimmune neuromyotonia (Isaacs' syndrome): an antibody-mediated potassium channelopathy". Ann. N. Y. Acad. Sci. 835 (1): 111–9. Bibcode:1997NYASA.835..111N. doi:10.1111/j.1749-6632.1997.tb48622.x. PMID 9616766.
- "Entrez Gene: KCNA1 potassium voltage-gated channel".
- "KCNA1 - Potassium voltage-gated channel subfamily A member 1 - Homo sapiens (Human) - KCNA1 gene & protein". www.uniprot.org.
- Bhalla T, Rosenthal JJ, Holmgren M, Reenan R (October 2004). "Control of human potassium channel inactivation by editing of a small mRNA hairpin". Nat. Struct. Mol. Biol. 11 (10): 950–6. doi:10.1038/nsmb825. PMID 15361858.
- Hoopengardner B, Bhalla T, Staber C, Reenan R (August 2003). "Nervous system targets of RNA editing identified by comparative genomics". Science. 301 (5634): 832–6. Bibcode:2003Sci...301..832H. doi:10.1126/science.1086763. PMID 12907802.
- Bhalla, Tarun; Rosenthal, Joshua J C; Holmgren, Miguel; Reenan, Robert (2004). "Control of human potassium channel inactivation by editing of a small mRNA hairpin". Nature Structural & Molecular Biology. 11 (10): 950–956. doi:10.1038/nsmb825. ISSN 1545-9993. PMID 15361858.
- Bezanilla, Francisco (2004). "RNA editing of a human potassium channel modifies its inactivation". Nature Structural & Molecular Biology. 11 (10): 915–916. doi:10.1038/nsmb1004-915. ISSN 1545-9993. PMID 15452561.
Further reading
- Grunnet M, Rasmussen HB, Hay-Schmidt A, et al. (2003). "KCNE4 is an inhibitory subunit to Kv1.1 and Kv1.3 potassium channels". Biophys. J. 85 (3): 1525–37. Bibcode:2003BpJ....85.1525G. doi:10.1016/S0006-3495(03)74585-8. PMC 1303329. PMID 12944270.
- Nie DY, Zhou ZH, Ang BT, et al. (2003). "Nogo-A at CNS paranodes is a ligand of Caspr: possible regulation of K(+) channel localization". EMBO J. 22 (21): 5666–78. doi:10.1093/emboj/cdg570. PMC 275427. PMID 14592966.
- Imbrici P, Cusimano A, D'Adamo MC, et al. (2003). "Functional characterization of an episodic ataxia type-1 mutation occurring in the S1 segment of hKv1.1 channels". Pflügers Arch. 446 (3): 373–9. doi:10.1007/s00424-002-0962-2. PMID 12799903.
- Glaudemans B, van der Wijst J, Scola RH, et al. (2009). "A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia". J. Clin. Invest. 119 (4): 936–42. doi:10.1172/JCI36948. PMC 2662556. PMID 19307729.
- Shook SJ, Mamsa H, Jen JC, et al. (2008). "Novel mutation in KCNA1 causes episodic ataxia with paroxysmal dyspnea". Muscle Nerve. 37 (3): 399–402. doi:10.1002/mus.20904. PMID 17912752.
- Gubitosi-Klug RA, Mancuso DJ, Gross RW (2005). "The human Kv1.1 channel is palmitoylated, modulating voltage sensing: Identification of a palmitoylation consensus sequence". Proc. Natl. Acad. Sci. U.S.A. 102 (17): 5964–8. Bibcode:2005PNAS..102.5964G. doi:10.1073/pnas.0501999102. PMC 1087951. PMID 15837928.
- Zhang ZH, Rhodes KJ, Childers WE, et al. (2004). "Disinactivation of N-type inactivation of voltage-gated K channels by an erbstatin analogue". J. Biol. Chem. 279 (28): 29226–30. doi:10.1074/jbc.M403290200. PMID 15136567.
- Kimura K, Wakamatsu A, Suzuki Y, et al. (2006). "Diversification of transcriptional modulation: large-scale identification and characterization of putative alternative promoters of human genes". Genome Res. 16 (1): 55–65. doi:10.1101/gr.4039406. PMC 1356129. PMID 16344560.
- Jow F, Zhang ZH, Kopsco DC, et al. (2004). "Functional coupling of intracellular calcium and inactivation of voltage-gated Kv1.1/Kvbeta1.1 A-type K+ channels". Proc. Natl. Acad. Sci. U.S.A. 101 (43): 15535–40. Bibcode:2004PNAS..10115535J. doi:10.1073/pnas.0402081101. PMC 524431. PMID 15486093.
- Imbrici P, Grottesi A, D'Adamo MC, et al. (2009). "Contribution of the central hydrophobic residue in the PXP motif of voltage-dependent K+ channels to S6 flexibility and gating properties". Channels (Austin). 3 (1): 39–45. doi:10.4161/chan.3.1.7548. PMID 19202350.
- Kinali M, Jungbluth H, Eunson LH, et al. (2004). "Expanding the phenotype of potassium channelopathy: severe neuromyotonia and skeletal deformities without prominent Episodic Ataxia". Neuromuscul. Disord. 14 (10): 689–93. doi:10.1016/j.nmd.2004.06.007. PMID 15351427.
- Demos MK, Macri V, Farrell K, et al. (2009). "A novel KCNA1 mutation associated with global delay and persistent cerebellar dysfunction". Mov. Disord. 24 (5): 778–82. doi:10.1002/mds.22467. PMID 19205071.
- Imbrici P, Gualandi F, D'Adamo MC, et al. (2008). "A novel KCNA1 mutation identified in an Italian family affected by episodic ataxia type 1". Neuroscience. 157 (3): 577–87. doi:10.1016/j.neuroscience.2008.09.022. PMID 18926884.
- Tan KM, Lennon VA, Klein CJ, et al. (2008). "Clinical spectrum of voltage-gated potassium channel autoimmunity". Neurology. 70 (20): 1883–90. doi:10.1212/01.wnl.0000312275.04260.a0. PMID 18474843.
- Chen H, von Hehn C, Kaczmarek LK, et al. (2007). "Functional analysis of a novel potassium channel (KCNA1) mutation in hereditary myokymia". Neurogenetics. 8 (2): 131–5. doi:10.1007/s10048-006-0071-z. PMC 1820748. PMID 17136396.
- Strausberg RL, Feingold EA, Grouse LH, et al. (2002). "Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences". Proc. Natl. Acad. Sci. U.S.A. 99 (26): 16899–903. Bibcode:2002PNAS...9916899M. doi:10.1073/pnas.242603899. PMC 139241. PMID 12477932.
- Gutman GA, Chandy KG, Grissmer S, et al. (2005). "International Union of Pharmacology. LIII. Nomenclature and molecular relationships of voltage-gated potassium channels". Pharmacol. Rev. 57 (4): 473–508. doi:10.1124/pr.57.4.10. PMID 16382104.
- Lee H, Wang H, Jen JC, et al. (2004). "A novel mutation in KCNA1 causes episodic ataxia without myokymia". Hum. Mutat. 24 (6): 536. doi:10.1002/humu.9295. PMID 15532032.
- Gu C, Jan YN, Jan LY (2003). "A conserved domain in axonal targeting of Kv1 (Shaker) voltage-gated potassium channels". Science. 301 (5633): 646–9. Bibcode:2003Sci...301..646G. doi:10.1126/science.1086998. PMID 12893943.
External links
- GeneReviews/NCBI/NIH/UW entry on Episodic Ataxia Type 1, Episodic Ataxia with Myokymia, Hereditary Cerebellar Ataxia with Neuromyotonia
- Kv1.1+Potassium+Channel at the US National Library of Medicine Medical Subject Headings (MeSH)
- KCNA1+protein,+human at the US National Library of Medicine Medical Subject Headings (MeSH)
PDB gallery | |
|---|---|
|