Episodic ataxia
Episodic ataxia (EA) is an autosomal dominant disorder characterized by sporadic bouts of ataxia (severe discoordination) with or without myokymia (continuous muscle movement). There are seven types recognized but the majority are due to two recognized entities.[1] Ataxia can be provoked by psychological stress or startle, or heavy exertion, including exercise. Symptoms can first appear in infancy. There are at least six loci for EA, of which 4 are known genes. Some patients with EA also have migraine or progressive cerebellar degenerative disorders, symptomatic of either familial hemiplegic migraine or spinocerebellar ataxia. Some patients respond to acetazolamide though others do not.
| Episodic ataxia | |
|---|---|
| Specialty | Neurology |
Signs and symptoms
Typically, episodic ataxia presents as bouts of ataxia induced by startle, stress, or exertion. Some patients also have continuous tremors of various motor groups, known as myokymia. Other patients have nystagmus, vertigo, tinnitus, diplopia or seizures.
Cause
The various symptoms of EA are caused by dysfunction of differing areas. Ataxia, the most common symptom, is due to misfiring of Purkinje cells in the cerebellum. This is either due to direct malfunction of these cells, such as in EA2, or improper regulation of these cells, such as in EA1. Seizures are likely due to altered firing of hippocampal neurons (KCNA1 null mice have seizures for this reason).
Pathophysiology
EA1: KCNA1
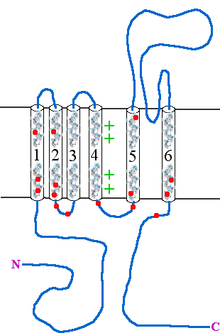
Type 1 episodic ataxia (EA1) is characterized by attacks of generalized ataxia induced by emotion or stress, with myokymia both during and between attacks. This disorder is also known as episodic ataxia with myokymia (EAM), hereditary paroxysmal ataxia with neuromyotonia and Isaacs-Mertens syndrome. Onset of EA1 occurs during early childhood to adolescence and persists throughout the patient's life. Attacks last from seconds to minutes. Mutations of the gene KCNA1, which encodes the voltage-gated potassium channel KV1.1, are responsible for this subtype of episodic ataxia. KV1.1 is expressed heavily in basket cells and interneurons that form GABAergic synapses on Purkinje cells. The channels aid in the repolarization phase of action potentials, thus affecting inhibitory input into Purkinje cells and, thereby, all motor output from the cerebellum. EA1 is an example of a synaptopathy. There are currently 17 KV1.1 mutations associated with EA1, Table 1 and Figure 1. 15 of these mutations have been at least partly characterized in cell culture based electrophysiological assays wherein 14 of these 15 mutations have demonstrated drastic alterations in channel function. As described in Table 1, most of the known EA1 associated mutations result in a drastic decrease in the amount of current through KV1.1 channels. Furthermore, these channels tend to activate at more positive potentials and slower rates, demonstrated by positive shifts in their V½ values and slower τ activation time constants, respectively. Some of these mutations, moreover, produce channels that deactivate at faster rates (deactivation τ), which would also result in decreased current through these channels. While these biophysical changes in channel properties likely underlie some of the decrease in current observed in experiments, many mutations also seem to result in misfolded or otherwise mistrafficked channels, which is likely to be the major cause of dysfunction and disease pathogenesis. It is assumed, though not yet proven, that decrease in KV1.1 mediated current leads to prolonged action potentials in interneurons and basket cells. As these channels are important in the regulation of Purkinje cell activity, it is likely that this results increased and aberrant inhibitory input into Purkinje cells and, thus, disrupted Purkinje cell firing and cerebellum output.
| Mutation | Position | Current amplitude (% wild-type) |
Activation | Deactivation (τ) | Other | References | |
|---|---|---|---|---|---|---|---|
| V½ | τ | ||||||
| V174F | S1 | 7.6% | 25mV positive | Unchanged | Unchanged | [2],[3],[4],[5] | |
| I177N | S1 | 5.9% | 60mV positive | Slower | Faster | Shorter mean open time and smaller single channel conductance | [6],[7] |
| F184C | S1 | 15.1% | 24mV positive | Slower | Slower | Fewer channels at membrane | [3],[4],[5],[8] |
| T226A | S2 | 5% | 15mV positive | Slower | Slower | [6],[9] | |
| T226M | S2 | 5% | 15mV positive | Slower | Slower | [5],[10] | |
| T226R | S2 | 3% | ? | ? | ? | [11] | |
| R239S | S2 | 0% | NA | NA | NA | Improper trafficking | [2],[4],[9] |
| A242P | S2 | 10% | 4mV Negative | Slower | Slower | [12] | |
| P244H | S2-3 | Unchanged | Unchanged | Unchanged | Unchanged | [12] | |
| F249I | S2-3 | 1% | Unchanged | Unchanged | Slower | Improper trafficking | [2],[4],[5] |
| G311S | S3-4 | 22.9% | 30mV positive | Unchanged | Unchanged | [9] | |
| E325D | S5 | 7.7% | 52.4mV positive | Faster | Faster | Impaired translation or stability | [3],[4],[5],[13],[14],[15] |
| L329I | S5 | ? | ? | ? | ? | [16] | |
| S342I | S5 | ? | ? | ? | ? | [17] | |
| V404I | S6 | Unchanged | 12mV positive | Slower | Slower | [6],[12] | |
| V408A | C-terminus | 68% | Unchanged | Faster | Faster | Shorter mean open time, more and larger sIPSCs in Mice | [2],[4],[5],[8],[13],[14],[15],[18] |
| R417X | C-terminus | 2% | 9mV positive | Slower | Faster | Misfolds and form membranous aggregates | [12],[19] |
| Current amplitude refers to the amount of current through mutant versus wild-type channels in cell culture or oocyte assays. Activation V½ is the potential at which the population of channels is half maximally activated which the accompanying τ is the time constant of the populations activation. Deactivation τ is similar to that of activation, referring instead to the time constant of population closing. sIPSCs are spontaneous inhibitory post synaptic currents. Cells with a red background indicate that this property will result in decreased KV1.1 current while cells with a green background indicate increased current through this channel. | |||||||
EA2: CACNA1A
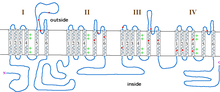
Type 2 episodic ataxia (EA2) is characterized by acetazolamide-responsive attacks of ataxia with or without migraine. Patients with EA2 may also present with progressive cerebellar atrophy, nystagmus, vertigo, visual disturbances and dysarthria. These symptoms last from hours to days, in contrast with EA1, which lasts from seconds to minutes. Attacks can be accompanied by increased heart rate and blood pressure, moderate to severe shaking, and stuttering. Like EA1, attacks can be precipitated by exercise, emotional stress/agitation, physical stress, or heat (overheated body temperature) but also by coffee and alcohol. EA2 is caused by mutations in CACNA1A, which encodes the P/Q-type voltage-gated calcium channel CaV2.1, and is also the gene responsible for causing spinocerebellar ataxia type-6 and familial hemiplegic migraine type-1. EA2 is also referred to as episodic ataxia with nystagmus, hereditary paroxysmal cerebellopathy, familial paroxysmal ataxia and acetazolamide-responsive hereditary paroxysmal cerebellar ataxia (AHPCA). There are currently 19 mutations associated with EA2, though only 3 have been characterized electrophysiologically, table 2 and figure 2. Of these, all result in decreased current through these channels. It is assumed that the other mutations, especially the splicing and frameshift mutations, also result in a drastic decrease in CaV2.1 currents, though this may not be the case for all mutations. CACNA1A is heavily expressed in Purkinje cells of the cerebellum where it is involved in coupling action potentials with neurotransmitter release. Thus, decrease in Ca2+ entry through CaV2.1 channels is expected to result in decreased output from Purkinje cells, even though they will fire at an appropriate rate. The tottering mouse is a widely used model to study EA2, as it developed a spontaneous homologous mutation in Cacna1a in the early 1960s.[20] Alternatively, some CACNA1A mutations, such as those seen in familial hemiplegic migraine type-1, result in increased Ca2+ entry and, thereby, aberrant transmitter release. This can also result in excitotoxicity, as may occur in some cases of spinocerebellar ataxia type-6.
| Mutation | Position | Effect | Cerebellar Signs | References |
|---|---|---|---|---|
| H253Y | D1-pore | ? | Yes | [21] |
| C271Y* | D1-pore | Decreased maximal current due to protein instability | Yes | [22] |
| G293R* | D1-pore | Decreased maximal current due to protein instability | Yes | [22],[23] |
| F624LfsX657 | D2S5 | ? | Yes | [21] |
| Q681RfsX780 | D2-pore | ? | Yes | [21] |
| S753fsX780 | D2S6 | ? | Yes | [24] |
| P1266LfsX1293 | D3S1 | ? | Yes | [24],[25] |
| R1278X | D3S1-2 | ? | Yes | [26] |
| F1391LfsX1429 | D3S5 | ? | Yes | [21] |
| Y1443X | D3-pore | ? | Yes | [24] |
| F1490K | D3S6 | No current, though expressed | Yes | [27] |
| R1546X | D4S1 | ? | Yes | [24] |
| A1593_Y1594delinsD | D4S2 | ? | Yes | [24] |
| R1661H | D4S4 | ? | Yes | [28] |
| R1664Q* | D4S4 | ? | Yes | [29] |
| E1756K | D4-pore | ? | Yes | [30] |
| Splicing | Intron 11 | ? | Yes | [24] |
| Splicing | Intron 26 | ? | Yes | [24] |
| Splicing | Intron 28 | ? | Yes | [25] |
* |
Also diagnosed as Spinocerebellar ataxia type-6 | |||
EA3: 1q42
Episodic ataxia type-3 (EA3) is similar to EA1 but often also presents with tinnitus and vertigo. Patients typically present with bouts of ataxia lasting less than 30 minutes and occurring once or twice daily. During attacks, they also have vertigo, nausea, vomiting, tinnitus and diplopia. These attacks are sometimes accompanied by headaches and precipitated by stress, fatigue, movement and arousal after sleep. Attacks generally begin in early childhood and last throughout the patients' lifetime. Acetazolamide administration has proved successful in some patients.[31] As EA3 is extremely rare, there is currently no known causative gene. The locus for this disorder has been mapped to the long arm of chromosome 1 (1q42).[32]
EA4
Also known as periodic vestibulocerebellar ataxia, type-4 episodic ataxia (EA4) is an extremely rare form of episodic ataxia differentiated from other forms by onset in the third to sixth generation of life, defective smooth pursuit and gaze-evoked nystagmus. Patients also present with vertigo and ataxia. There are only two known families with EA4, both located in North Carolina. The locus for EA4 is unknown.
EA5: CACNB4
There are two known families with type-5 episodic ataxia (EA5).
These patients can present with an overlapping phenotype of ataxia and seizures similar to juvenile myoclonic epilepsy. In fact, juvenile myoclonic epilepsy and EA5 are allelic and produce proteins with similar dysfunction.
Patients with pure EA5 present with recurrent episodes of ataxia with vertigo. Between attacks they have nystagmus and dysarthria. These patients are responsive to acetazolamide.
Both juvenile myoclonic epilepsy and EA5 are a result of mutations in CACNB4, a gene that encodes the calcium channel β4 subunit. This subunit coassembles with α-subunits and produces channels that slowly inactivate after opening.
EA5 patients have a cysteine to phenylalanine mutation at position 104.
Thus results in channels with 30% greater current than wild-type.
As this subunit is expressed in the cerebellum, it is assumed that such increased current results in neuronal hyperexcitability
Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia.
EA6: SLC1A3
Type-6 episodic ataxia (EA6) is a rare form of episodic ataxia, identified initially in a 10-year-old boy who first presented with 30 minute bouts of decreased muscle tone during infancy. He required "balance therapy" as a young child to aid in walking and has a number of ataxic attacks, each separated by months to years. These attacks were precipitated by fever. He has cerebellar atrophy and subclinical seizures. During later attacks, he also presented with distortions of the left hemifield, ataxia, slurred speech, followed by headache. After enrolling in school, he developed bouts of rhythmic arm jerking with concomitant confusion, also lasting approximately 30 minutes. He also has presented, at various times, with migraines. This patient carries a proline to arginine substitution in the fifth transmembrane-spanning segment of the gene SLC1A3. This gene encodes the excitatory amino acid transporter 1 (EAAT1) protein, which is responsible for glutamate uptake. In cell culture assays, this mutation results in drastically decreased glutamate uptake in a dominant-negative manner. This is likely due to decreased synthesis or protein stability. As this protein is expressed heavily in the brainstem and cerebellum, it is likely that this mutation results in excitotoxicity and/or hyperexcitability leading to ataxia and seizures.[33] Mutations in EAAT1 (GLAST) have subsequently been identified in a family with episodic ataxia.[34]
Diagnosis
Treatment
Depending on subtype, many patients find that acetazolamide therapy is useful in preventing attacks. In some cases, persistent attacks result in tendon shortening, for which surgery is required.
References
- Riant F, Vahedi K, Tournier-Lasserve E (2011) Hereditary episodic ataxia. Rev Neurol (Paris)
- Browne D, Gancher S, Nutt J, Brunt E, Smith E, Kramer P, Litt M (1994). "Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene, KCNA1". Nat Genet. 8 (2): 136–40. doi:10.1038/ng1094-136. PMID 7842011.
- Browne D, Brunt E, Griggs R, Nutt J, Gancher S, Smith E, Litt M (1995). "Identification of two new KCNA1 mutations in episodic ataxia/myokymia families". Hum Mol Genet. 4 (9): 1671–2. doi:10.1093/hmg/4.9.1671. PMID 8541859.
- Adelman J, Bond C, Pessia M, Maylie J (1995). "Episodic ataxia results from voltage-dependent potassium channels with altered functions". Neuron. 15 (6): 1449–54. doi:10.1016/0896-6273(95)90022-5. PMID 8845167.
- Zerr P, Adelman J, Maylie J (1998). "Episodic ataxia mutations in Kv1.1 alter potassium channel function by dominant negative effects or haploinsufficiency". J Neurosci. 18 (8): 2842–8. doi:10.1523/JNEUROSCI.18-08-02842.1998. PMC 6792579. PMID 9526001.
- Scheffer H, Brunt E, Mol G, van der Vlies P, Stulp R, Verlind E, Mantel G, Averyanov Y, Hofstra R, Buys C (1998). "Three novel KCNA1 mutations in episodic ataxia type I families". Hum Genet. 102 (4): 464–6. doi:10.1007/s004390050722. PMID 9600245.
- Imbrici P, Cusimano A, D'Adamo M, De Curtis A, Pessia M (2003). "Functional characterization of an episodic ataxia type-1 mutation occurring in the S1 segment of hKv1.1 channels". Pflügers Arch. 446 (3): 373–9. doi:10.1007/s00424-002-0962-2. PMID 12799903.
- Bretschneider F, Wrisch A, Lehmann-Horn F, Grissmer S (1999). "Expression in mammalian cells and electrophysiological characterization of two mutant Kv1.1 channels causing episodic ataxia type 1 (EA-1)". Eur J Neurosci. 11 (7): 2403–12. doi:10.1046/j.1460-9568.1999.00659.x. PMID 10383630.
- Zerr P, Adelman J, Maylie J (1998). "Characterization of three episodic ataxia mutations in the human Kv1.1 potassium channel". FEBS Lett. 431 (3): 461–4. doi:10.1016/S0014-5793(98)00814-X. PMID 9714564.
- Comu S, Giuliani M, Narayanan V (1996). "Episodic ataxia and myokymia syndrome: a new mutation of potassium channel gene Kv1.1". Ann Neurol. 40 (4): 684–7. doi:10.1002/ana.410400422. PMID 8871592.
- Zuberi S, Eunson L, Spauschus A, De Silva R, Tolmie J, Wood N, McWilliam R, Stephenson J, Kullmann D, Hanna M (1999). "A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy". Brain. 122 (5): 817–25. doi:10.1093/brain/122.5.817. PMID 10355668.
- Eunson L, Rea R, Zuberi S, Youroukos S, Panayiotopoulos C, Liguori R, Avoni P, McWilliam R, Stephenson J, Hanna M, Kullmann D, Spauschus A (2000). "Clinical, genetic, and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability". Ann Neurol. 48 (4): 647–56. doi:10.1002/1531-8249(200010)48:4<647::AID-ANA12>3.0.CO;2-Q. PMID 11026449.
- D'Adamo M, Liu Z, Adelman J, Maylie J, Pessia M (1998). "Episodic ataxia type-1 mutations in the hKv1.1 cytoplasmic pore region alter the gating properties of the channel". EMBO J. 17 (5): 1200–7. doi:10.1093/emboj/17.5.1200. PMC 1170468. PMID 9482717.
- D'Adamo M, Imbrici P, Sponcichetti F, Pessia M (1999). "Mutations in the KCNA1 gene associated with episodic ataxia type-1 syndrome impair heteromeric voltage-gated K(+) channel function". FASEB J. 13 (11): 1335–45. doi:10.1096/fasebj.13.11.1335. PMID 10428758.
- Maylie B, Bissonnette E, Virk M, Adelman J, Maylie J (2002). "Episodic ataxia type 1 mutations in the human Kv1.1 potassium channel alter hKvbeta 1-induced N-type inactivation". J Neurosci. 22 (12): 4786–93. doi:10.1523/JNEUROSCI.22-12-04786.2002. PMC 6757728. PMID 12077175.
- Knight M, Storey E, McKinlay Gardner R, Hand P, Forrest S (2000). "Identification of a novel missense mutation L329I in the episodic ataxia type 1 gene KCNA1--a challenging problem". Hum Mutat. 16 (4): 374. doi:10.1002/1098-1004(200010)16:4<374::AID-HUMU15>3.0.CO;2-4. PMID 11013453.
- Lee H, Wang H, Jen J, Sabatti C, Baloh R, Nelson S (2004). "A novel mutation in KCNA1 causes episodic ataxia without myokymia". Hum Mutat. 24 (6): 536. doi:10.1002/humu.9295. PMID 15532032.
- Herson P, Virk M, Rustay N, Bond C, Crabbe J, Adelman J, Maylie J (2003). "A mouse model of episodic ataxia type-1". Nat Neurosci. 6 (4): 378–83. doi:10.1038/nn1025. PMID 12612586.
- Manganas L, Akhtar S, Antonucci D, Campomanes C, Dolly J, Trimmer J (2001). "Episodic ataxia type-1 mutations in the Kv1.1 potassium channel display distinct folding and intracellular trafficking properties". J Biol Chem. 276 (52): 49427–34. doi:10.1074/jbc.M109325200. PMID 11679591.
- Green, M. C.; Sidman, R. L. (September 1962). "Tottering--a neuromusclar mutation in the mouse. And its linkage with oligosyndacylism". The Journal of Heredity. 53 (5): 233–237. doi:10.1093/oxfordjournals.jhered.a107180. ISSN 0022-1503. PMID 13950100.
- van den Maagdenberg A, Kors E, Brunt E, van Paesschen W, Pascual J, Ravine D, Keeling S, Vanmolkot K, Vermeulen F, Terwindt G, Haan J, Frants R, Ferrari M (2002). "Episodic ataxia type 2. Three novel truncating mutations and one novel missense mutation in the CACNA1A gene". J Neurol. 249 (11): 1515–9. doi:10.1007/s00415-002-0860-8. PMID 12420090.
- Wan J, Khanna R, Sandusky M, Papazian D, Jen J, Baloh R (2005). "CACNA1A mutations causing episodic and progressive ataxia alter channel trafficking and kinetics". Neurology. 64 (12): 2090–7. doi:10.1212/01.WNL.0000167409.59089.C0. PMID 15985579.
- Yue Q, Jen J, Nelson S, Baloh R (1997). "Progressive ataxia due to a missense mutation in a calcium-channel gene". Am J Hum Genet. 61 (5): 1078–87. doi:10.1086/301613. PMC 1716037. PMID 9345107.
- Denier C, Ducros A, Vahedi K, Joutel A, Thierry P, Ritz A, Castelnovo G, Deonna T, Gérard P, Devoize J, Gayou A, Perrouty B, Soisson T, Autret A, Warter J, Vighetto A, Van Bogaert P, Alamowitch S, Roullet E, Tournier-Lasserve E (1999). "High prevalence of CACNA1A truncations and broader clinical spectrum in episodic ataxia type 2". Neurology. 52 (9): 1816–21. doi:10.1212/WNL.52.9.1816. PMID 10371528.
- Ophoff R, Terwindt G, Vergouwe M, van Eijk R, Oefner P, Hoffman S, Lamerdin J, Mohrenweiser H, Bulman D, Ferrari M, Haan J, Lindhout D, van Ommen G, Hofker M, Ferrari M, Frants R (1996). "Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4". Cell. 87 (3): 543–52. doi:10.1016/S0092-8674(00)81373-2. PMID 8898206.
- Yue Q, Jen J, Thwe M, Nelson S, Baloh R (1998). "De novo mutation in CACNA1A caused acetazolamide-responsive episodic ataxia". Am J Med Genet. 77 (4): 298–301. doi:10.1002/(SICI)1096-8628(19980526)77:4<298::AID-AJMG9>3.0.CO;2-J. PMID 9600739.
- Guida S, Trettel F, Pagnutti S, Mantuano E, Tottene A, Veneziano L, Fellin T, Spadaro M, Stauderman K, Williams M, Volsen S, Ophoff R, Frants R, Jodice C, Frontali M, Pietrobon D (2001). "Complete loss of P/Q calcium channel activity caused by a CACNA1A missense mutation carried by patients with episodic ataxia type 2". Am J Hum Genet. 68 (3): 759–64. doi:10.1086/318804. PMC 1274487. PMID 11179022.
- Friend K, Crimmins D, Phan T, Sue C, Colley A, Fung V, Morris J, Sutherland G, Richards R (1999). "Detection of a novel missense mutation and second recurrent mutation in the CACNA1A gene in individuals with EA-2 and FHM". Hum Genet. 105 (3): 261–5. doi:10.1007/s004390051099. PMID 10987655.
- Tonelli A, D'Angelo M, Salati R, Villa L, Germinasi C, Frattini T, Meola G, Turconi A, Bresolin N, Bassi M (2006). "Early onset, non fluctuating spinocerebellar ataxia and a novel missense mutation in CACNA1A gene". J Neurol Sci. 241 (1–2): 13–7. doi:10.1016/j.jns.2005.10.007. PMID 16325861.
- Denier C, Ducros A, Durr A, Eymard B, Chassande B, Tournier-Lasserve E (2001). "Missense CACNA1A mutation causing episodic ataxia type 2". Arch Neurol. 58 (2): 292–5. doi:10.1001/archneur.58.2.292. PMID 11176968.
- Steckley J, Ebers G, Cader M, McLachlan R (2001). "An autosomal dominant disorder with episodic ataxia, vertigo, and tinnitus". Neurology. 57 (8): 1499–502. doi:10.1212/wnl.57.8.1499. PMID 11673600.
- Cader M, Steckley J, Dyment D, McLachlan R, Ebers G (2005). "A genome-wide screen and linkage mapping for a large pedigree with episodic ataxia". Neurology. 65 (1): 156–8. doi:10.1212/01.wnl.0000167186.05465.7c. PMID 16009908.
- Jen J, Wan J, Palos T, Howard B, Baloh R (2005). "Mutation in the glutamate transporter EAAT1 causes episodic ataxia, hemiplegia, and seizures". Neurology. 65 (4): 529–34. doi:10.1212/01.WNL.0000172638.58172.5a. PMID 16116111.
- de Vries B, Mamsa H, Stam AH, et al. (2009). "Episodic ataxia associated with EAAT1 mutation C186S affecting glutamate reuptake". Arch. Neurol. 66 (1): 97–101. doi:10.1001/archneurol.2008.535. PMID 19139306.
External links
| Classification | |
|---|---|
| External resources |