Ethinylestradiol
Ethinylestradiol (EE) is an estrogen medication which is used widely in birth control pills in combination with progestins.[8][9] In the past, EE was widely used for various indications such as the treatment of menopausal symptoms, gynecological disorders, and certain hormone-sensitive cancers. It is usually taken by mouth but is also used as a patch and vaginal ring.[8][12]
 | |
 | |
| Clinical data | |
|---|---|
| Pronunciation | /ˌɛθɪnɪlˌiːstrəˈdaɪ.əl/ |
| Trade names | Numerous |
| Other names | Ethynylestradiol; Ethinyl estradiol; Ethinyl oestradiol; EE; EE2; 17α-Ethynylestradiol; 17α-Ethynylestra-1,3,5(10)-triene-3,17β-diol; NSC-10973[1] |
| AHFS/Drugs.com | International Drug Names |
| MedlinePlus | a604032 |
| License data | |
| Pregnancy category |
|
| Routes of administration | • By mouth (tablet) • Transdermal (patch) • Vaginal (ring) |
| Drug class | Estrogen |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 38–48%[2][3][4] |
| Protein binding | 97–98% (to albumin;[5] is not bound to SHBG)[6] |
| Metabolism | Liver (primarily CYP3A4)[7] |
| Metabolites | • Ethinylestradiol sulfate[8][9] • Others[8][9] |
| Elimination half-life | 7–36 hours[7][2][10][11] |
| Excretion | Feces: 62%[10] Urine: 38%[10] |
| Identifiers | |
| |
| CAS Number | |
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| KEGG | |
| ChEBI | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| ECHA InfoCard | 100.000.311 |
| Chemical and physical data | |
| Formula | C20H24O2 |
| Molar mass | 296.410 g·mol−1 |
| 3D model (JSmol) | |
| Melting point | 182 to 184 °C (360 to 363 °F) |
| |
| |
| (verify) | |
The general side effects of EE include breast tenderness and enlargement, headache, fluid retention, and nausea among others.[8] In men, EE can additionally cause breast development, feminization in general, hypogonadism, and sexual dysfunction. Rare but serious side effects include blood clots, liver damage, and cancer of the uterus.[8]
EE is an estrogen, or an agonist of the estrogen receptors, the biological target of estrogens like estradiol.[8] It is a synthetic derivative of estradiol, a natural estrogen, and differs from it in various ways.[8] Compared to estradiol, EE has greatly improved bioavailability when taken by mouth, is more resistant to metabolism, and shows relatively increased effects in certain parts of the body like the liver and uterus.[8] These differences make EE more favorable for use in birth control pills than estradiol, though also result in an increased risk of blood clots and certain other rare adverse effects.[8]
EE was developed in the 1930s and was introduced for medical use in 1943.[13][14] The medication started being used in birth control pills in the 1960s.[15] Today, EE is found in almost all combined forms of birth control pills and is nearly the exclusive estrogen used for this purpose, making it one of if not the most widely used estrogens.[16][17]
Medical uses
There are many uses for EE. It is most commonly used as contraception in combined oral contraceptives (COC), also known as birth control, to prevent pregnancy after sex. EE in its birth control formulation is not only used to prevent pregnancy, but can also be used to treat absence of menstruation, symptoms during menstruation, and acne.
EE is also used as menopausal hormone therapy. The main reason for using HRT in menopausal women is to relieve common vasomotor symptoms such as hot flashes, night sweats, and flushing. Studies have found that estrogen replacement helps improve these symptoms when compared to a placebo.[18] Other common menopause symptoms such as vaginal dryness (which can cause pain during sexual intercourse), vaginal itching, and depressed mood, can benefit from HRT. In addition to treatment of menopausal symptoms, EE has been used as a component of feminizing hormone therapy for transgender women.[19] However, it is no longer commonly used nor recommended for this purpose, with estradiol having largely superseded it.[19]
EE can also be used to treat hypogonadism in women, prevent osteoporosis in women, and has been used as palliative care for prostate cancer in men and breast cancer in women.[9][20]
EE or any estrogen alone is contraindicated for women who have a uterus due to the increased risk of endometrial cancer; giving a progestogen with an estrogen mitigates the risk.[21]
| Route/form | Estrogen | Dosage | |
|---|---|---|---|
| Oral | Estradiol | 10 mg 3x/day AI-resistant: 2 mg 1–3x/day | |
| Estradiol valerate | AI-resistant: 2 mg 1–3x/day | ||
| Conjugated estrogens | 10 mg 3x/day | ||
| Ethinylestradiol | 0.5–1 mg 3x/day | ||
| Diethylstilbestrol | 5 mg 3x/day | ||
| Dienestrol | 5 mg 3x/day | ||
| Dimestrol | 30 mg/day | ||
| Chlorotrianisene | 24 mg/day | ||
| IM or SC injection | Estradiol benzoate | 5 mg 2–3x/week | |
| Estradiol dipropionate | 5 mg 2–3x/week | ||
| Estradiol valerate | 30 mg 1x/2 weeks | ||
| Polyestradiol phosphate | 40–80 mg 1x/4 weeks | ||
| Estrone | 5 mg ≥3x/week | ||
| Notes: (1) Only in women who are at least 5 years postmenopausal. (2) Dosages are not necessarily equivalent. Sources: See template. | |||
| Route/form | Estrogen | Dosage | |
|---|---|---|---|
| Oral | Estradiol | 1–2 mg 3x/day | |
| Conjugated estrogens | 1.25–2.5 mg 3x/day | ||
| Ethinylestradiol | 0.15–3 mg/day | ||
| Ethinylestradiol sulfonate | 1–2 mg 1x/week | ||
| Diethylstilbestrol | 1–3 mg/day | ||
| Dienestrol | 5 mg/day | ||
| Hexestrol | 5 mg/day | ||
| Fosfestrol | 100–480 mg 1–3x/day | ||
| Chlorotrianisene | 12–48 mg/day | ||
| Quadrosilan | 900 mg/day | ||
| Estramustine phosphate | 140–1400 mg/day | ||
| Transdermal patch | Estradiol | 2–6x 100 μg/day Scrotal: 1x 100 μg/day | |
| IM or SC injection | Estradiol benzoate | 1.66 mg 3x/week | |
| Estradiol dipropionate | 5 mg 1x/week | ||
| Estradiol valerate | 10–40 mg 1x/1–2 weeks | ||
| Estradiol undecylate | 100 mg 1x/4 weeks | ||
| Polyestradiol phosphate | Alone: 160–320 mg 1x/4 weeks With oral EE: 40–80 mg 1x/4 weeks | ||
| Estrone | 2–4 mg 2–3x/week | ||
| IV injection | Fosfestrol | 300–1200 mg 1–7x/week | |
| Estramustine phosphate | 240–450 mg/day | ||
| Note: Dosages are not necessarily equivalent. Sources: See template. | |||
Available forms
EE is available in combination with a progestin in a vast number of COCs.[22] It is also available in combination with progestins as a transdermal contraceptive patch and as a contraceptive vaginal ring.[12] In addition, there is a single preparation containing very low doses of EE (2.5 and 5 µg) plus a progestin in an oral tablet that remains in use for menopausal hormone therapy.[12] EE was previously available by itself under the brand name Estinyl in the form of 0.02, 0.05, and 0.5 mg (20, 50, and 500 µg) tablets.[23]
The amount of EE in COCs has reduced over the years.[9] Previously, COCs contained high doses of EE of as much as 100 µg/day.[24] Doses of more than 50 µg EE are considered high-dose, doses of 30 and 35 µg EE are considered low-dose, and doses of 10 to 25 µg EE are considered very low dose.[25] Today, COCs generally contain 10 to 50 µg EE.[25] The higher doses of EE were discontinued due to a high risk of VTE and cardiovascular problems.[24]
Contraindications
EE should be avoided in individuals with a history of or known susceptibility to arterial or venous thrombosis (blood clots), due to an increased risk of cardiovascular problems such as venous thromboembolism (VTE), myocardial infarction, and ischemic stroke.[26] This includes women with:
- History of deep vein thrombosis (DVT) or pulmonary embolism (PE) not receiving anticoagulants
- Acute DVT/PE
- Prolonged immobilization due to major surgery
- Advanced diabetes mellitus with vascular disease
- Migraine with aura
- Hypertension ≥160/100
- Vascular disease
- Current and history of ischemic heart disease
- Multiple risk factors for atherosclerotic cardiovascular disease (e.g. older age, smoking, diabetes, hypertension, low HDL, high LDL, or high triglyceride levels)
- Age ≥35 and smoking ≥15 cigarettes/day
- History of cerebrovascular accident
- Systemic lupus erythematosus with positive (or unknown) antiphospholipid antibodies
- Complicated valvular heart disease
Except when being used to treat it, EE should be avoided in women with current breast cancer due to a possible worsening of prognosis.[27]
EE should also be avoided in breastfeeding women who are less than 21 days postpartum due to an increased risk of VTE.[28] EE use in breastfeeding women who are at least 21 days postpartum should be discussed with a provider and include information on the advantages, disadvantages, and alternatives for using EE.[28]
Due to risk of cholestatic hepatotoxicity, it is widely considered that COCs containing EE should be avoided in women with a history of cholestasis of pregnancy, hepatic tumors, active hepatitis, and familial defects in biliary excretion.[29]
Side effects
The severity of side effects can vary based on the dose and administration route of EE.[30] General side effects of EE are the same as for other estrogens and include breast tenderness, headache, fluid retention (bloating), nausea, dizziness, and weight gain.[10][29] The estrogen component of oral contraceptives, which is almost always EE, can cause breast tenderness and fullness.[23] In males, EE has additional side effects, including gynecomastia (breast development), feminization in general, hypogonadism, infertility, and sexual dysfunction (e.g., reduced libido and erectile dysfunction). In men who received high-dose estrogen therapy with 200 μg/day oral EE for more than three months, gynecomastia occurred in 98% and decreased libido occurred in 42 to 73%.[31]
| Beneficial effects | Adverse effects | ||
|---|---|---|---|
| Disease | RR | Disease | RR |
| Iron-deficiency anemia | 0.58 | Cardiovascular diseases (total) | 1.5 |
| Menorrhagia | 0.52 | Myocardial infarction (heart attack) (total) | 3.3 |
| Irregular menstruation | 0.65 | Myocardial infarction (non-smokers) | 1.0 |
| Intermenstrual bleeding | 0.72 | Myocardial infarction (light smokers) | 3.5 |
| Dysmenorrhea | 0.37 | Myocardial infarction (heavy smokers) | 20 |
| Pelvic inflammatory disease (incidence) | 0.50 | Cerebrovascular diseases (total) | 1.4 |
| Pelvic inflammatory disease (hospitalization) | 0.22 | Cerebral thromboses (strokes) | 2.5 |
| Trichomonas vaginitis | 0.56 | Subarachnoidal bleeding (heavy smokers) | 10 |
| Benign breast disease | 0.69 | Pulmonary embolism | 3.0 |
| Fibrocystic breast disease | 0.66 | Deep vein thromboses | 2.5 |
| Benign breast fibroadenomas | 0.35 | Gall-bladder diseases | 3.0 |
| Rheumatoid arthritis | 0.49 | Benign liver tumors | 50 |
| Endometrial cancer | 0.40–0.50 | Hepatocellular carcinoma | 3.0 |
| Ovarian cancer (incidence) | 0.37–0.64 | Erythema nodosum et multiforme | 3.0 |
| Ovarian cancer (death) | 0.20 | Pruritus (itching) | 2.0 |
| Benign follicular cysts (high-dose COCs) | 0.24 | Photosensitive eczema | 4.0 |
| Acne vulgaris | 0.44 | Irritant agent eczema | 2.0 |
| Low bone mineral density (later in life) | 0.35a | Dermatitis (eczema) | 2.0 |
| Ectopic pregnancy | 0.19 | Chloasma (melasma) | 1.5 |
| Cervicitis (6 years of use) | 3.0 | ||
| Chlamydia infections | 2.5 | ||
| Footnotes: a = Odds ratio. Sources: See template. | |||
Long-term effects
Blood clots
VTE is a blood clot in a vein, and includes deep vein thrombosis (DVT) and pulmonary embolism (PE).[8][32][33] Estrogens are known to increase the risk of VTE due to their effects on liver synthesis of coagulation factors.[8][32][33] EE carries a greater risk of blood clot formation and VTE than does natural estradiol, which is thought to be due to structural differences between the two compounds and different susceptibilities to liver inactivation.[8]
A 2012 meta-analysis estimated that the absolute risk of VTE is 2 per 10,000 women for non-use, 8 per 10,000 women for EE and levonorgestrel-containing birth control pills, and 10 to 15 per 10,000 women for birth control pills containing EE and a third- or fourth-generation progestin such as desogestrel or drospirenone.[34] For comparison, the absolute risk of VTE is generally estimated as 1 to 5 per 10,000 woman–years for non-use, 5 to 20 per 10,000 woman–years for pregnancy, and 40 to 65 per 10,000 woman–years for the postpartum period.[34] Modern COCs are associated with about a 2- to 4-fold higher risk of VTE than non-use.[34] The route of administration of EE does not appear to influence VTE risk, as EE/progestin-containing contraceptive vaginal rings and contraceptive patches have the same or even higher risk of VTE than COCs.[34][35] Pregnancy is associated with about a 4.3-fold increase in risk of VTE.[34] It has been estimated that at least 300 to 400 healthy young women die each year in the United States due to VTE caused by EE-containing birth control pills.[36]
Modern COCs contain 10 to 35 μg EE, but typically 20, 30, or 35 μg.[34][37] The initial formulations of COCs that were introduced in the 1960s contained 100 to 150 μg EE.[38][39][37] However, it was soon found that EE is associated with increased risk of VTE and that the risk is dose-dependent.[37] Following these events, the dose of EE was greatly reduced, and is now always less than 50 μg.[23][40][41] These lower doses have a significantly reduced risk of VTE with no loss of contraceptive effectiveness.[37] Gerstman et al. (1991) found that COCs containing more than 50 μg EE had 1.7-fold and COCs containing 50 μg EE 1.5-fold the risk of VTE of COCs containing less than 50 μg.[42] A 2014 Cochrane review found that COCs containing 50 μg EE with levonorgestrel had 2.1- to 2.3-fold the risk of COCs containing 30 μg or 20 μg EE with levonorgestrel, respectively.[34] COCs containing 20 μg EE are likewise associated with a significantly lower risk of cardiovascular events than COCs containing 30 or 40 μg EE.[43] However, discontinuation of COCs is more common with doses of EE from 10 to 20 μg due to problematic changes in bleeding patterns.[44]
Women with thrombophilia have a dramatically higher risk of VTE with EE-containing contraception than women without thrombophilia.[34][35] Depending on the condition, risk of VTE can be increased 5- to 50-fold relative to non-use in such women.[34][35]
Sex hormone-binding globulin (SHBG) levels indicate hepatic estrogenic exposure and may be a surrogate marker for coagulation and VTE risk with estrogen therapy, although this topic has been debated.[45][46][47] SHBG levels with birth control pills containing different progestins are increased by 1.5 to 2-fold with levonorgestrel, 2.5- to 4-fold with desogestrel and gestodene, 3.5- to 4-fold with drospirenone and dienogest, and 4- to 5-fold with cyproterone acetate.[45] Contraceptive vaginal rings and contraceptive patches likewise have been found to increase SHBG levels by 2.5-fold and 3.5-fold, respectively.[45] Birth control pills containing high doses of ethinylestradiol (>50 μg) can increase SHBG levels by 5- to 10-fold, which is similar to the increase that occurs during pregnancy.[48] Conversely, increases in SHBG levels are much lower with estradiol, especially when used parenterally.[49][50][51][52][53] High-dose parenteral polyestradiol phosphate therapy has been found to increase SHBG levels by about 1.5-fold.[52][52]
| Ethinylestradiol dose | No. of VTE cases | Woman-years | VTE rate | Adjusted RRa |
|---|---|---|---|---|
| Low (<50 μg) | 53 | 127,000 | 4.2 in 10,000 woman-years | 1.0 |
| Intermediate (50 μg) | 69 | 98,000 | 7.0 in 10,000 woman-years | 1.5 |
| High (>50 μg) | 20 | 20,000 | 10.0 in 10,000 woman-years | 1.7 |
| All | 142 | 245,000 | 5.8 in 10,000 woman-years | – |
| Footnotes: a = Relative to low-dose (not to non-use). Notes: In birth control pills containing a first-generation progestin, such as norethisterone or levonorgestrel. Sources: Main: [42][54] Additional: [39] | ||||
| Type | Route | Medications | Odds ratio (95% CI) |
|---|---|---|---|
| Menopausal hormone therapy | Oral | Estradiol alone ≤1 mg/day >1 mg/day | 1.27 (1.16–1.39)* 1.22 (1.09–1.37)* 1.35 (1.18–1.55)* |
| Conjugated estrogens alone ≤0.625 mg/day >0.625 mg/day | 1.49 (1.39–1.60)* 1.40 (1.28–1.53)* 1.71 (1.51–1.93)* | ||
| Estradiol/medroxyprogesterone acetate | 1.44 (1.09–1.89)* | ||
| Estradiol/dydrogesterone ≤1 mg/day E2 >1 mg/day E2 | 1.18 (0.98–1.42) 1.12 (0.90–1.40) 1.34 (0.94–1.90) | ||
| Estradiol/norethisterone ≤1 mg/day E2 >1 mg/day E2 | 1.68 (1.57–1.80)* 1.38 (1.23–1.56)* 1.84 (1.69–2.00)* | ||
| Estradiol/norgestrel or estradiol/drospirenone | 1.42 (1.00–2.03) | ||
| Conjugated estrogens/medroxyprogesterone acetate | 2.10 (1.92–2.31)* | ||
| Conjugated estrogens/norgestrel ≤0.625 mg/day CEEs >0.625 mg/day CEEs | 1.73 (1.57–1.91)* 1.53 (1.36–1.72)* 2.38 (1.99–2.85)* | ||
| Tibolone alone | 1.02 (0.90–1.15) | ||
| Raloxifene alone | 1.49 (1.24–1.79)* | ||
| Transdermal | Estradiol alone ≤50 μg/day >50 μg/day | 0.96 (0.88–1.04) 0.94 (0.85–1.03) 1.05 (0.88–1.24) | |
| Conjugated estrogens alone | 1.04 (0.76–1.43) | ||
| Estradiol/progestogen | 0.88 (0.73–1.01) | ||
| Vaginal | Estradiol alone | 0.84 (0.73–0.97) | |
| Combined birth control | Oral | Ethinylestradiol/norethisterone | 2.56 (2.15–3.06)* |
| Ethinylestradiol/levonorgestrel | 2.38 (2.18–2.59)* | ||
| Ethinylestradiol/norgestimate | 2.53 (2.17–2.96)* | ||
| Ethinylestradiol/desogestrel | 4.28 (3.66–5.01)* | ||
| Ethinylestradiol/gestodene | 3.64 (3.00–4.43)* | ||
| Ethinylestradiol/drospirenone | 4.12 (3.43–4.96)* | ||
| Ethinylestradiol/cyproterone acetate | 4.27 (3.57–5.11)* | ||
| Notes: (1) Nested case–control studies (2015, 2019) based on data from the QResearch and Clinical Practice Research Datalink (CPRD) databases. (2) Bioidentical progesterone was not included, but is known to be associated with no additional risk relative to estrogen alone. Footnotes: * = Statistically significant (p < 0.01). Sources: See template. | |||
Cardiovascular issues
When used orally at high dosages, for instance as a form of high-dose estrogen therapy in men with prostate cancer and in women with breast cancer, synthetic and non-bioidentical estrogens like EE and diethylstilbestrol are associated with fairly high rates of severe cardiovascular complications such as VTE, myocardial infarction, and stroke.[20][55][56] Diethylstilbestrol has been associated with an up to 35% risk of cardiovascular toxicity and death and a 15% incidence of VTE in men treated with it for prostate cancer.[55][56] EE has a to some degree lower risk of cardiovascular complications than does diethylstilbestrol when used in the treatment of prostate cancer in men.[9] However, both EE and diethylstilbestrol nonetheless have highly disproportionate effects on liver protein synthesis, which is thought to be responsible for their cardiovascular toxicity.[8][56]
In contrast to oral synthetic estrogens like EE and diethylstilbestrol, high-dosage polyestradiol phosphate and transdermal estradiol have not been found to increase the risk of cardiovascular mortality or thromboembolism in men with prostate cancer.[56][57][58] However, significantly increased cardiovascular morbidity has been observed with high-dosage polyestradiol phosphate.[56][57][58] In any case, these estrogens are considered to be much safer than oral synthetic estrogens like EE and diethylstilbestrol.[56][57][58] In addition, ethinylestradiol sulfonate (EES), an oral but parenteral-like long-lasting prodrug of EE, is used in the treatment of prostate cancer, and is said to have a considerably better profile of cardiovascular safety than EE.[9]
Because of its disproportionate effects on liver protein synthesis and associated cardiovascular risks, synthetic estrogens like EE and diethylstilbestrol are no longer used in menopausal hormone therapy.[9] They are also being replaced by parenteral forms of estradiol like polyestradiol phosphate and transdermal estradiol in the treatment of prostate cancer.[56]
Liver damage
EE has rarely (at the low dosages that are now used in COCs) been associated with cholestatic hepatotoxicity similarly to 17α-alkylated androgens/anabolic steroids and 17α-ethynylated 19-nortestosterone progestins.[59][60] Glucuronide metabolites of EE, via effects on the ABCB11 (BSEP) and MRP2 (ABCC2) proteins and consequent changes in bile flow and bile salt excretion, appear to be responsible for the cholestasis.[61] High concentrations of estradiol, via its metabolite estradiol glucuronide, are also implicated in cholestasis, for instance in cholestasis of pregnancy.[60] However, the incidence and severity of cholestatic hepatotoxicity appear to be much greater with EE than with estradiol, which is due to its 17α-ethynyl substitution and consequent reduced metabolism.[29]
Uterine cancer
The high doses of EE that were used in early COCs were associated with a significantly increased risk of endometrial cancer in certain preparations, for instance those containing the progestogen dimethisterone.[62] Unopposed estrogens like EE have carcinogenic effects in the endometrium and progestogens protect against these effects, but dimethisterone is a relatively weak progestogen and was unable to adequately antagonize the endometrial carcinogenic effects of EE, in turn resulting in the increased risk of endometrial cancer.[62] COCs containing dimethisterone have since been discontinued (with more potent progestogens used instead) and doses of EE in COCs in general have been dramatically reduced, abrogating the risk.[62] In turn, most studies of modern COCs have found a decreased risk of endometrial cancer.[63]
Ecological Effects
Wastewater contains various estrogens, including EE, that are not completely broken down by wastewater treatment procedures.[64] The input of artificial estrogens into freshwater ecosystems affects fish and amphibian populations. Chronic exposure to low levels of EE over seven years led to the collapse of fathead minnow populations in an experimental lake in Ontario, Canada.[64] EE changed oogenesis in female fish and feminized male fish such that they produced a protein associated with egg maturation, vitellogenin, as well as early-stage eggs.[64] In amphibians, exposure to EE can reduce hatching success and alter gonadal development.[65] Exposure to hormones can change frogs' gonadal development even though it is encoded in their genes.[65] A study of mink frogs found more intersex tadpoles in those experimentally exposed to EE than those not exposed to EE, and green frogs showed much lower rates of hatching success.[65]
Overdose
Estrogens like EE are relatively safe in acute overdose.
Interactions
EE is metabolized by certain cytochrome P450 isoforms, including CYP3A4 and CYP2C9.[66] Thus, inducers of enzymes such as CYP3A4 can decrease circulating concentrations of EE.[29] Examples of inducers include anticonvulsants like phenytoin, primidone, ethosuximide, phenobarbital, and carbamazepine; azole antifungals like fluconazole; and rifamycin antibiotics like rifampin (rifampicin).[29] Conversely, inhibitors of CYP3A4 and other cytochrome P450 enzymes may increase circulating levels of EE.[29] An example is troleandomycin, which is a potent and highly selective inhibitor of CYP3A4.[29]
Paracetamol (acetaminophen) has been found to competitively inhibit the sulfation of EE, with pretreatment of 1,000 mg of paracetamol significantly increasing the AUC levels of EE (by 22%) and decreasing the AUC levels of ethinylestradiol sulfate (EE sulfate) in women.[29] The same has been found for ascorbic acid (vitamin C) and EE, although the significance of the interaction has been regarded as dubious.[29]
In contrast to estradiol, it is unlikely that there is a pharmacokinetic interaction between smoking (which potently induces certain cytochrome P450 enzymes and markedly increases the 2-hydroxylation of estradiol) and EE.[29] This suggests that estradiol and EE are metabolized by different cytochrome P450 enzymes.[29] There is, however, an increased risk of cardiovascular complications with smoking and EE, similarly to the case of smoking and other estrogens.[29]
The 19-nortestosterone progestins, gestodene and, to a lesser extent, desogestrel, have been found to inhibit cytochrome P450 enzymes and to progressively inhibit the metabolism and increase the concentrations of EE.[29]
EE has been found to significantly increase (by 38%) the AUC of omeprazole (which is metabolized by CYP2C19).[29]
Pharmacology
Pharmacodynamics
EE is an estrogen similarly to natural estrogens like estradiol and conjugated estrogens (Premarin) and synthetic estrogens like diethylstilbestrol. It binds to and activates both isoforms of the estrogen receptor, ERα and ERβ.[9] In one study, EE was found to have 233% and 38% of the affinity of estradiol for the ERα and ERβ, respectively.[67] In another study, it was found to possess 194% and 151% of the affinity of estradiol for the ERα and ERβ, respectively.[68] EE also appears to act as a potent agonist of the G protein-coupled estrogen receptor (GPER) (affinity unknown), a membrane estrogen receptor, similarly to estradiol.[69][70][71][72] Estrogens have antigonadotropic effects through activation of the ERα.[73] As a contraceptive, EE acts in concert with a progestin to inhibit the mid-cycle surge in luteinizing hormone (LH) and follicle-stimulating hormone (FSH) via its antigonadotropic effects, thereby inhibiting folliculogenesis and preventing ovulation and hence the possibility of pregnancy.[74][75]
EE is a long-acting estrogen, with a nuclear retention of about 24 hours.[54]
Orally, EE is on the order of 100 times as potent by weight as natural estrogens like micronized estradiol and conjugated estrogens, which is largely due to substantially greater resistance to first-pass metabolism.[76][77][78] It is specifically in the range of 80 to 200 times as potent as estropipate (piperazine estrone sulfate), which has similar potency to micronized estradiol, in terms of systemic estrogenic potency.[79][80] In contrast, the potencies of EE and natural estrogens are similar when they are administered intravenously, due to the bypassing of first-pass metabolism.[37] Relative to its prodrug mestranol, EE is about 1.7 times as potent by weight orally.[77]
| Ligand | Other names | Relative binding affinities (RBA, %)a | Absolute binding affinities (Ki, nM)a | Action | ||
|---|---|---|---|---|---|---|
| ERα | ERβ | ERα | ERβ | |||
| Estradiol | E2; 17β-Estradiol | 100 | 100 | 0.115 (0.04–0.24) | 0.15 (0.10–2.08) | Estrogen |
| Estrone | E1; 17-Ketoestradiol | 16.39 (0.7–60) | 6.5 (1.36–52) | 0.445 (0.3–1.01) | 1.75 (0.35–9.24) | Estrogen |
| Estriol | E3; 16α-OH-17β-E2 | 12.65 (4.03–56) | 26 (14.0–44.6) | 0.45 (0.35–1.4) | 0.7 (0.63–0.7) | Estrogen |
| Estetrol | E4; 15α,16α-Di-OH-17β-E2 | 4.0 | 3.0 | 4.9 | 19 | Estrogen |
| Alfatradiol | 17α-Estradiol | 20.5 (7–80.1) | 8.195 (2–42) | 0.2–0.52 | 0.43–1.2 | Metabolite |
| 16-Epiestriol | 16β-Hydroxy-17β-estradiol | 7.795 (4.94–63) | 50 | ? | ? | Metabolite |
| 17-Epiestriol | 16α-Hydroxy-17α-estradiol | 55.45 (29–103) | 79–80 | ? | ? | Metabolite |
| 16,17-Epiestriol | 16β-Hydroxy-17α-estradiol | 1.0 | 13 | ? | ? | Metabolite |
| 2-Hydroxyestradiol | 2-OH-E2 | 22 (7–81) | 11–35 | 2.5 | 1.3 | Metabolite |
| 2-Methoxyestradiol | 2-MeO-E2 | 0.0027–2.0 | 1.0 | ? | ? | Metabolite |
| 4-Hydroxyestradiol | 4-OH-E2 | 13 (8–70) | 7–56 | 1.0 | 1.9 | Metabolite |
| 4-Methoxyestradiol | 4-MeO-E2 | 2.0 | 1.0 | ? | ? | Metabolite |
| 2-Hydroxyestrone | 2-OH-E1 | 2.0–4.0 | 0.2–0.4 | ? | ? | Metabolite |
| 2-Methoxyestrone | 2-MeO-E1 | <0.001–<1 | <1 | ? | ? | Metabolite |
| 4-Hydroxyestrone | 4-OH-E1 | 1.0–2.0 | 1.0 | ? | ? | Metabolite |
| 4-Methoxyestrone | 4-MeO-E1 | <1 | <1 | ? | ? | Metabolite |
| 16α-Hydroxyestrone | 16α-OH-E1; 17-Ketoestriol | 2.0–6.5 | 35 | ? | ? | Metabolite |
| 2-Hydroxyestriol | 2-OH-E3 | 2.0 | 1.0 | ? | ? | Metabolite |
| 4-Methoxyestriol | 4-MeO-E3 | 1.0 | 1.0 | ? | ? | Metabolite |
| Estradiol sulfate | E2S; Estradiol 3-sulfate | <1 | <1 | ? | ? | Metabolite |
| Estradiol disulfate | Estradiol 3,17β-disulfate | 0.0004 | ? | ? | ? | Metabolite |
| Estradiol 3-glucuronide | E2-3G | 0.0079 | ? | ? | ? | Metabolite |
| Estradiol 17β-glucuronide | E2-17G | 0.0015 | ? | ? | ? | Metabolite |
| Estradiol 3-gluc. 17β-sulfate | E2-3G-17S | 0.0001 | ? | ? | ? | Metabolite |
| Estrone sulfate | E1S; Estrone 3-sulfate | <1 | <1 | >10 | >10 | Metabolite |
| Estradiol benzoate | EB; Estradiol 3-benzoate | 10 | ? | ? | ? | Estrogen |
| Estradiol 17β-benzoate | E2-17B | 11.3 | 32.6 | ? | ? | Estrogen |
| Estrone methyl ether | Estrone 3-methyl ether | 0.145 | ? | ? | ? | Estrogen |
| ent-Estradiol | 1-Estradiol | 1.31–12.34 | 9.44–80.07 | ? | ? | Estrogen |
| Equilin | 7-Dehydroestrone | 13 (4.0–28.9) | 13.0–49 | 0.79 | 0.36 | Estrogen |
| Equilenin | 6,8-Didehydroestrone | 2.0–15 | 7.0–20 | 0.64 | 0.62 | Estrogen |
| 17β-Dihydroequilin | 7-Dehydro-17β-estradiol | 7.9–113 | 7.9–108 | 0.09 | 0.17 | Estrogen |
| 17α-Dihydroequilin | 7-Dehydro-17α-estradiol | 18.6 (18–41) | 14–32 | 0.24 | 0.57 | Estrogen |
| 17β-Dihydroequilenin | 6,8-Didehydro-17β-estradiol | 35–68 | 90–100 | 0.15 | 0.20 | Estrogen |
| 17α-Dihydroequilenin | 6,8-Didehydro-17α-estradiol | 20 | 49 | 0.50 | 0.37 | Estrogen |
| Δ8-Estradiol | 8,9-Dehydro-17β-estradiol | 68 | 72 | 0.15 | 0.25 | Estrogen |
| Δ8-Estrone | 8,9-Dehydroestrone | 19 | 32 | 0.52 | 0.57 | Estrogen |
| Ethinylestradiol | EE; 17α-Ethynyl-17β-E2 | 120.9 (68.8–480) | 44.4 (2.0–144) | 0.02–0.05 | 0.29–0.81 | Estrogen |
| Mestranol | EE 3-methyl ether | ? | 2.5 | ? | ? | Estrogen |
| Moxestrol | RU-2858; 11β-Methoxy-EE | 35–43 | 5–20 | 0.5 | 2.6 | Estrogen |
| Methylestradiol | 17α-Methyl-17β-estradiol | 70 | 44 | ? | ? | Estrogen |
| Diethylstilbestrol | DES; Stilbestrol | 129.5 (89.1–468) | 219.63 (61.2–295) | 0.04 | 0.05 | Estrogen |
| Hexestrol | Dihydrodiethylstilbestrol | 153.6 (31–302) | 60–234 | 0.06 | 0.06 | Estrogen |
| Dienestrol | Dehydrostilbestrol | 37 (20.4–223) | 56–404 | 0.05 | 0.03 | Estrogen |
| Benzestrol (B2) | – | 114 | ? | ? | ? | Estrogen |
| Chlorotrianisene | TACE | 1.74 | ? | 15.30 | ? | Estrogen |
| Triphenylethylene | TPE | 0.074 | ? | ? | ? | Estrogen |
| Triphenylbromoethylene | TPBE | 2.69 | ? | ? | ? | Estrogen |
| Tamoxifen | ICI-46,474 | 3 (0.1–47) | 3.33 (0.28–6) | 3.4–9.69 | 2.5 | SERM |
| Afimoxifene | 4-Hydroxytamoxifen; 4-OHT | 100.1 (1.7–257) | 10 (0.98–339) | 2.3 (0.1–3.61) | 0.04–4.8 | SERM |
| Toremifene | 4-Chlorotamoxifen; 4-CT | ? | ? | 7.14–20.3 | 15.4 | SERM |
| Clomifene | MRL-41 | 25 (19.2–37.2) | 12 | 0.9 | 1.2 | SERM |
| Cyclofenil | F-6066; Sexovid | 151–152 | 243 | ? | ? | SERM |
| Nafoxidine | U-11,000A | 30.9–44 | 16 | 0.3 | 0.8 | SERM |
| Raloxifene | – | 41.2 (7.8–69) | 5.34 (0.54–16) | 0.188–0.52 | 20.2 | SERM |
| Arzoxifene | LY-353,381 | ? | ? | 0.179 | ? | SERM |
| Lasofoxifene | CP-336,156 | 10.2–166 | 19.0 | 0.229 | ? | SERM |
| Ormeloxifene | Centchroman | ? | ? | 0.313 | ? | SERM |
| Levormeloxifene | 6720-CDRI; NNC-460,020 | 1.55 | 1.88 | ? | ? | SERM |
| Ospemifene | Deaminohydroxytoremifene | 2.63 | 1.22 | ? | ? | SERM |
| Bazedoxifene | – | ? | ? | 0.053 | ? | SERM |
| Etacstil | GW-5638 | 4.30 | 11.5 | ? | ? | SERM |
| ICI-164,384 | – | 63.5 (3.70–97.7) | 166 | 0.2 | 0.08 | Antiestrogen |
| Fulvestrant | ICI-182,780 | 43.5 (9.4–325) | 21.65 (2.05–40.5) | 0.42 | 1.3 | Antiestrogen |
| Propylpyrazoletriol | PPT | 49 (10.0–89.1) | 0.12 | 0.40 | 92.8 | ERα agonist |
| 16α-LE2 | 16α-Lactone-17β-estradiol | 14.6–57 | 0.089 | 0.27 | 131 | ERα agonist |
| 16α-Iodo-E2 | 16α-Iodo-17β-estradiol | 30.2 | 2.30 | ? | ? | ERα agonist |
| Methylpiperidinopyrazole | MPP | 11 | 0.05 | ? | ? | ERα antagonist |
| Diarylpropionitrile | DPN | 0.12–0.25 | 6.6–18 | 32.4 | 1.7 | ERβ agonist |
| 8β-VE2 | 8β-Vinyl-17β-estradiol | 0.35 | 22.0–83 | 12.9 | 0.50 | ERβ agonist |
| Prinaberel | ERB-041; WAY-202,041 | 0.27 | 67–72 | ? | ? | ERβ agonist |
| ERB-196 | WAY-202,196 | ? | 180 | ? | ? | ERβ agonist |
| Erteberel | SERBA-1; LY-500,307 | ? | ? | 2.68 | 0.19 | ERβ agonist |
| SERBA-2 | – | ? | ? | 14.5 | 1.54 | ERβ agonist |
| Coumestrol | – | 9.225 (0.0117–94) | 64.125 (0.41–185) | 0.14–80.0 | 0.07–27.0 | Xenoestrogen |
| Genistein | – | 0.445 (0.0012–16) | 33.42 (0.86–87) | 2.6–126 | 0.3–12.8 | Xenoestrogen |
| Equol | – | 0.2–0.287 | 0.85 (0.10–2.85) | ? | ? | Xenoestrogen |
| Daidzein | – | 0.07 (0.0018–9.3) | 0.7865 (0.04–17.1) | 2.0 | 85.3 | Xenoestrogen |
| Biochanin A | – | 0.04 (0.022–0.15) | 0.6225 (0.010–1.2) | 174 | 8.9 | Xenoestrogen |
| Kaempferol | – | 0.07 (0.029–0.10) | 2.2 (0.002–3.00) | ? | ? | Xenoestrogen |
| Naringenin | – | 0.0054 (<0.001–0.01) | 0.15 (0.11–0.33) | ? | ? | Xenoestrogen |
| 8-Prenylnaringenin | 8-PN | 4.4 | ? | ? | ? | Xenoestrogen |
| Quercetin | – | <0.001–0.01 | 0.002–0.040 | ? | ? | Xenoestrogen |
| Ipriflavone | – | <0.01 | <0.01 | ? | ? | Xenoestrogen |
| Miroestrol | – | 0.39 | ? | ? | ? | Xenoestrogen |
| Deoxymiroestrol | – | 2.0 | ? | ? | ? | Xenoestrogen |
| β-Sitosterol | – | <0.001–0.0875 | <0.001–0.016 | ? | ? | Xenoestrogen |
| Resveratrol | – | <0.001–0.0032 | ? | ? | ? | Xenoestrogen |
| α-Zearalenol | – | 48 (13–52.5) | ? | ? | ? | Xenoestrogen |
| β-Zearalenol | – | 0.6 (0.032–13) | ? | ? | ? | Xenoestrogen |
| Zeranol | α-Zearalanol | 48–111 | ? | ? | ? | Xenoestrogen |
| Taleranol | β-Zearalanol | 16 (13–17.8) | 14 | 0.8 | 0.9 | Xenoestrogen |
| Zearalenone | ZEN | 7.68 (2.04–28) | 9.45 (2.43–31.5) | ? | ? | Xenoestrogen |
| Zearalanone | ZAN | 0.51 | ? | ? | ? | Xenoestrogen |
| Bisphenol A | BPA | 0.0315 (0.008–1.0) | 0.135 (0.002–4.23) | 195 | 35 | Xenoestrogen |
| Endosulfan | EDS | <0.001–<0.01 | <0.01 | ? | ? | Xenoestrogen |
| Kepone | Chlordecone | 0.0069–0.2 | ? | ? | ? | Xenoestrogen |
| o,p'-DDT | – | 0.0073–0.4 | ? | ? | ? | Xenoestrogen |
| p,p'-DDT | – | 0.03 | ? | ? | ? | Xenoestrogen |
| Methoxychlor | p,p'-Dimethoxy-DDT | 0.01 (<0.001–0.02) | 0.01–0.13 | ? | ? | Xenoestrogen |
| HPTE | Hydroxychlor; p,p'-OH-DDT | 1.2–1.7 | ? | ? | ? | Xenoestrogen |
| Testosterone | T; 4-Androstenolone | <0.0001–<0.01 | <0.002–0.040 | >5000 | >5000 | Androgen |
| Dihydrotestosterone | DHT; 5α-Androstanolone | 0.01 (<0.001–0.05) | 0.0059–0.17 | 221–>5000 | 73–1688 | Androgen |
| Nandrolone | 19-Nortestosterone; 19-NT | 0.01 | 0.23 | 765 | 53 | Androgen |
| Dehydroepiandrosterone | DHEA; Prasterone | 0.038 (<0.001–0.04) | 0.019–0.07 | 245–1053 | 163–515 | Androgen |
| 5-Androstenediol | A5; Androstenediol | 6 | 17 | 3.6 | 0.9 | Androgen |
| 4-Androstenediol | – | 0.5 | 0.6 | 23 | 19 | Androgen |
| 4-Androstenedione | A4; Androstenedione | <0.01 | <0.01 | >10000 | >10000 | Androgen |
| 3α-Androstanediol | 3α-Adiol | 0.07 | 0.3 | 260 | 48 | Androgen |
| 3β-Androstanediol | 3β-Adiol | 3 | 7 | 6 | 2 | Androgen |
| Androstanedione | 5α-Androstanedione | <0.01 | <0.01 | >10000 | >10000 | Androgen |
| Etiocholanedione | 5β-Androstanedione | <0.01 | <0.01 | >10000 | >10000 | Androgen |
| Methyltestosterone | 17α-Methyltestosterone | <0.0001 | ? | ? | ? | Androgen |
| Ethinyl-3α-androstanediol | 17α-Ethynyl-3α-adiol | 4.0 | <0.07 | ? | ? | Estrogen |
| Ethinyl-3β-androstanediol | 17α-Ethynyl-3β-adiol | 50 | 5.6 | ? | ? | Estrogen |
| Progesterone | P4; 4-Pregnenedione | <0.001–0.6 | <0.001–0.010 | ? | ? | Progestogen |
| Norethisterone | NET; 17α-Ethynyl-19-NT | 0.085 (0.0015–<0.1) | 0.1 (0.01–0.3) | 152 | 1084 | Progestogen |
| Norethynodrel | 5(10)-Norethisterone | 0.5 (0.3–0.7) | <0.1–0.22 | 14 | 53 | Progestogen |
| Tibolone | 7α-Methylnorethynodrel | 0.5 (0.45–2.0) | 0.2–0.076 | ? | ? | Progestogen |
| Δ4-Tibolone | 7α-Methylnorethisterone | 0.069–<0.1 | 0.027–<0.1 | ? | ? | Progestogen |
| 3α-Hydroxytibolone | – | 2.5 (1.06–5.0) | 0.6–0.8 | ? | ? | Progestogen |
| 3β-Hydroxytibolone | – | 1.6 (0.75–1.9) | 0.070–0.1 | ? | ? | Progestogen |
| Footnotes: a = (1) Binding affinity values are of the format "median (range)" (# (#–#)), "range" (#–#), or "value" (#) depending on the values available. The full sets of values within the ranges can be found in the Wiki code. (2) Binding affinities were determined via displacement studies in a variety of in-vitro systems with labeled estradiol and human ERα and ERβ proteins (except the ERβ values from Kuiper et al. (1997), which are rat ERβ). Sources: See template page. | ||||||
| Estrogen | Relative binding affinities (%) | ||||||
|---|---|---|---|---|---|---|---|
| ER | AR | PR | GR | MR | SHBG | CBG | |
| Estradiol | 100 | 7.9 | 2.6 | 0.6 | 0.13 | 8.7–12 | <0.1 |
| Estradiol benzoate | ? | ? | ? | ? | ? | <0.1–0.16 | <0.1 |
| Estradiol valerate | 2 | ? | ? | ? | ? | ? | ? |
| Estrone | 11–35 | <1 | <1 | <1 | <1 | 2.7 | <0.1 |
| Estrone sulfate | 2 | 2 | ? | ? | ? | ? | ? |
| Estriol | 10–15 | <1 | <1 | <1 | <1 | <0.1 | <0.1 |
| Equilin | 40 | ? | ? | ? | ? | ? | 0 |
| Alfatradiol | 15 | <1 | <1 | <1 | <1 | ? | ? |
| Epiestriol | 20 | <1 | <1 | <1 | <1 | ? | ? |
| Ethinylestradiol | 100–112 | 1–3 | 15–25 | 1–3 | <1 | 0.18 | <0.1 |
| Mestranol | 1 | ? | ? | ? | ? | <0.1 | <0.1 |
| Methylestradiol | 67 | 1–3 | 3–25 | 1–3 | <1 | ? | ? |
| Moxestrol | 12 | <0.1 | 0.8 | 3.2 | <0.1 | <0.2 | <0.1 |
| Diethylstilbestrol | ? | ? | ? | ? | ? | <0.1 | <0.1 |
| Notes: Reference ligands (100%) were progesterone for the PR, testosterone for the AR, estradiol for the ER, dexamethasone for the GR, aldosterone for the MR, dihydrotestosterone for SHBG, and cortisol for CBG. Sources: See template. | |||||||
| Estrogen | Type | HF | VE | UCa | FSH | LH | HDL-C | SHBG | CBG | AGT | Liver |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Estradiol | Bioidentical | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 | 1.0 |
| Estrone | Bioidentical | ? | ? | ? | 0.3 | 0.3 | ? | ? | ? | ? | ? |
| Estriol | Bioidentical | 0.3 | 0.3 | 0.1 | 0.3 | 0.3 | 0.2 | ? | ? | ? | 0.67 |
| Estrone sulfate | Bioidentical | ? | 0.9 | 0.9 | 0.8–0.9 | 0.9 | 0.5 | 0.9 | 0.5–0.7 | 1.4–1.5 | 0.56–1.7 |
| Conjugated estrogens | Natural | 1.2 | 1.5 | 2.0 | 1.1–1.3 | 1.0 | 1.5 | 3.0–3.2 | 1.3–1.5 | 5.0 | 1.3–4.5 |
| Equilin sulfate | Natural | ? | ? | 1.0 | ? | ? | 6.0 | 7.5 | 6.0 | 7.5 | ? |
| Ethinylestradiol | Synthetic | 120 | 150 | 400 | 60–150 | 100 | 400 | 500–600 | 500–600 | 350 | 2.9–5.0 |
| Diethylstilbestrol | Synthetic | ? | ? | ? | 2.9–3.4 | ? | ? | 26–28 | 25–37 | 20 | 5.7–7.5 |
| Notes: Values are ratios, with estradiol as standard (i.e., 1.0). Abbreviations: HF = Clinical relief of hot flashes. VE = Increased proliferation of vaginal epithelium. UCa = Decrease in UCa. FSH = Suppression of FSH levels. LH = Suppression of LH levels. HDL-C, SHBG, CBG, and AGT = Increase in the serum levels of these liver proteins. Liver = Ratio of liver estrogenic effects to general/systemic estrogenic effects (specifically hot flashes relief and gonadotropin suppression). Type: Bioidentical = Identical to those found in humans. Natural = Naturally occurring but not identical to those found in humans (e.g., estrogens of other species). Synthetic = Man-made, does not occur naturally in animals or in the environment. Sources: See template. | |||||||||||
| Estrogen | Type | Class | ETD (2–3 weeks) | EPD (2–3 weeks) | MSD (2–3 weeks) | MSD (daily) | OID (daily) | TSD (daily) |
|---|---|---|---|---|---|---|---|---|
| Estradiol (non-micronized) | Bioidentical | Steroidal | 30 mg | ≥120–300 mg | 120 mg | 6 mg | ? | ? |
| Estradiol (micronized) | Bioidentical | Steroidal | 6–12 mg | 60–80 mg | 14–42 mg | 1–2 mg | >5 mg | >8 mg |
| Estradiol valerate | Bioidentical | Steroidal | 6–12 mg | 60–80 mg | 14–42 mg | 1–2 mg | ? | >8 mg |
| Estradiol benzoate | Bioidentical | Steroidal | ? | 60–140 mg | ? | ? | ? | ? |
| Estriol | Bioidentical | Steroidal | 20 mga | 120–150 mgb | 28–126 mg | 1–6 mg | >5 mg | ? |
| Estriol succinate | Bioidentical | Steroidal | ? | 140–150 mgb | 28–126 mg | 2–6 mg | ? | ? |
| Estrone sulfate | Bioidentical | Steroidal | 12 mg | 60 mg | 42 mg | 2 mg | ? | ? |
| Conjugated estrogens | Natural | Steroidal | 5–12 mg | 60–80 mg | 8.4–25 mg | 0.625–1.25 mg | >3.75 mg | 7.5 mg |
| Ethinylestradiol | Synthetic | Steroidal | 200 μg | 1–2 mg | 280 μg | 20–40 μg | 100 μg | 100 μg |
| Mestranol | Synthetic | Steroidal | 300 μg | 1.5–3.0 mg | 300–600 μg | 25–30 μg | >80 μg | ? |
| Quinestrol | Synthetic | Steroidal | 300 μg | 2–4 mg | 500 μg | 25–50 μg | ? | ? |
| Methylestradiol | Synthetic | Steroidal | ? | 2 mg | ? | ? | ? | ? |
| Diethylstilbestrol | Synthetic | Nonsteroidal | 2.5 mg | 20–30 mg | 11 mg | 0.5–2.0 mg | >5 mg | 3 mg |
| Diethylstilbestrol dipropionate | Synthetic | Nonsteroidal | ? | 15–30 mg | ? | ? | ? | ? |
| Dienestrol | Synthetic | Nonsteroidal | 5 mg | 30–40 mg | 42 mg | 0.5–4.0 mg | ? | ? |
| Dienestrol diacetate | Synthetic | Nonsteroidal | 3–5 mg | 30–60 mg | ? | ? | ? | ? |
| Hexestrol | Synthetic | Nonsteroidal | ? | 70–110 mg | ? | ? | ? | ? |
| Chlorotrianisene | Synthetic | Nonsteroidal | ? | >100 mg | ? | ? | >48 mg | ? |
| Methallenestril | Synthetic | Nonsteroidal | ? | 400 mg | ? | ? | ? | ? |
| Footnotes: a = Very variable, often higher. b = In divided doses, 3x/day; irregular and atypical proliferation. Sources: See template. | ||||||||
Antiandrogenic and antigonadotropic effects
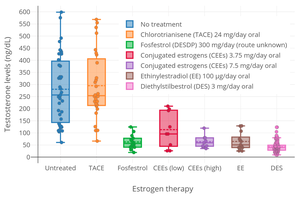
EE is a potent functional antiandrogen in both women and men.[82] It mediates its antiandrogenic effects by 1) stimulating the production of sex hormone-binding globulin (SHBG) in the liver, which decreases free and thus bioactive concentrations of testosterone in the blood; and by 2) suppressing luteinizing hormone (LH) secretion from the pituitary gland, which decreases production of testosterone by the gonads.[82][83][22][84] Birth control pills that contain EE are useful in the treatment of androgen-dependent conditions like acne and hirsutism by virtue of their antiandrogenic effects.[82][85]
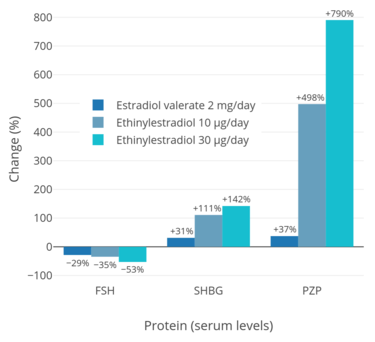
Birth control pills containing EE have been found to increase circulating SHBG levels by 2- to 4-fold in women and to reduce free testosterone concentrations by 40 to 80%.[22] Birth control pills containing high doses of EE can increase SHBG levels in women by as much as 5- to 10-fold.[48] This is similar to the 5- to 10-fold increase in SHBG levels that occurs during pregnancy.[48] Due to the marked increase in SHBG levels, free testosterone levels become very low during treatment with EE-containing birth control pills.[10] In men, a study found that treatment with a relatively low dosage of 20 μg/day EE for five weeks increased circulating SHBG levels by 150% and, due to the accompanying decrease in free testosterone levels, increased total circulating levels of testosterone by 50% (via upregulation of gonadal testosterone production due to reduced negative feedback by androgens on the hypothalamic–pituitary–gonadal axis).[83] The stimulation of hepatic SHBG production by EE is far stronger than with other estrogens like estradiol, owing to the high resistance of EE to inactivation in the liver and hence its disproportionate effects in this part of the body.[8][10][86]
Estrogens are antigonadotropins and are able to suppress the secretion of LH and FSH from the pituitary gland and by extension gonadal testosterone production.[87][88] High-dose estrogen therapy, including with EE, is able to suppress testosterone levels in men by around 95%, or into the castrate/female range.[89][87][88] The dosage of EE required for use as a component of hormone therapy for preoperative transgender women is 50 to 100 µg/day.[90] This high dosage is associated with a high incidence of VTE, particularly in those over the age of 40 years, and it has been said that it should not be used.[90] The dosage of EE used in the treatment of prostate cancer in men is 150 to 1,000 µg/day (0.15–1.0 mg/day).[9][91] A dosage of EE of 50 μg twice daily (100 μg/day total) has been found to suppress testosterone levels in men to an equivalent extent as 3 mg/day oral diethylstilbestrol, which is the minimum dosage of diethylstilbestrol required to consistently suppress testosterone levels into the castrate range.[92] The ovulation-inhibiting dose of EE by itself and not in combination with a progestin in women is 100 μg/day.[93][94] However, it has been found to be about 75 to 90% effective at inhibiting ovulation at a dosage of 20 μg/day and about 97 or 98% effective at a dosage of 50 μg/day.[95][96][97][98] In another study, ovulation occurred in 25.2% with an EE dose of 50 μg/day.[99]
Lower dosages of EE also have significant antigonadotropic effects.[90] A "very low" dosage of 15 µg/day EE has been described as the "borderline" amount required for suppression of LH and testosterone levels in men, and a study found that LH and testosterone levels were "reliably" suppressed in men by a dosage of 30 µg/day EE.[9] However, other clinical studies have found that 20 µg/day EE increased testosterone levels by 50% in men (as described above)[83] and that dosages of 32 µg/day and 42 µg/day EE suppressed FSH levels in men but did not significantly affect LH levels.[9] A stronger suppression of testosterone levels was observed in men following daily treatment with a combined oral contraceptive containing 50 µg ethinylestradiol and 0.5 mg norgestrel for 9 days.[9] However, investigation revealed that the progestin was the more important component responsible for the suppression in testosterone levels.[9] In accordance, the progestin component of COCs is primarily responsible for inhibition of ovulation in women.[9] A combination of 20 µg/day EE and 10 mg/day methyltestosterone was found to suppress FSH secretion in men to an extent sufficient to stop spermatogenesis.[9] Studies in women have found that 50 µg/day EE suppresses LH and FSH levels both by about 70% in postmenopausal women.[80]
In addition to its antigonadotropic effects, EE can significantly suppress androgen production by the adrenal glands at high concentrations.[9][100][101] One study found that treatment with a high dosage of 100 µg/day EE suppressed circulating adrenal androgen levels by 27 to 48% in transgender women.[9][100][101] This may additionally contribute to suppression of androgen levels by estrogens.[9][100][101]
Effects on liver protein synthesis
EE has marked effects on liver protein synthesis, even at low dosages and regardless of route of administration.[9][8] These effects are mediated by its estrogenic activity.[9][8] The medication dose-dependently increases circulating levels of SHBG, corticosteroid-binding globulin (CBG), and thyroxine-binding globulin (TBG), and also affects a broad range of other liver proteins.[9][8] EE affects triglyceride levels at a dose as low as 1 μg/day and LDL and HDL cholesterol levels at a dose as low as 2.5 μg/day.[102] EE affects several hepatic proteins at a dosage as low as 5 µg/day.[9] At doses above 20 µg/day, the incremental effects of EE on liver protein synthesis become continuously smaller.[9]
EE at 5 μg/day has been found to increase SHBG levels by 100% in postmenopausal women, while a dosage of 20 µg/day EE increased them by 200%.[9] Androgens decrease hepatic SHBG production, and have been found to oppose the effects of EE on SHBG levels.[9] This is of particular relevance when it is considered that many progestins used in COCs have varying degrees of weak androgenic activity.[9] A combination of 20 µg/day EE and 0.25 mg/day levonorgestrel, a progestin with relatively high androgenicity, decreases SHBG levels by 50%; 30 µg/day EE and 0.25 mg/day levonorgestrel has no effect on SHBG levels; 30 µg/day EE and 0.15 mg/day levonorgestrel increases SHBG levels by 30%; and triphasic COCs containing EE and levonorgestrel increase SHBG levels by 100 to 150%.[9] The combination of 30 µg/day EE and 150 µg/day desogestrel, a progestin with relatively weak androgenicity than levonorgestrel, increases SHBG levels by 200%, while the combination of 35 µg/day EE and 2 mg/day cyproterone acetate, a progestin with potent antiandrogenic activity, increases SHBG levels by 400%.[9] As such, the type and dosage of progestin contained in COCs potently moderates the effects of EE on SHBG levels.[9]
A dosage of 10 µg/day EE has been found to increase CBG levels by 50%, while a dosage of 20 µg/day EE increased them by 100%.[9] Progestins that are progesterone derivatives have no effect on CBG levels, while androgenic progestins like the 19-nortestosterone derivatives have only a weak effect on CBG levels.[9] COCs may increase CBG levels by 100 to 150%.[9] A dosage of 5 µg/day EE has been found to increase TBG levels by 40%, while a dosage of 20 µg/day EE increased them by 60%.[9] Progestins that are progesterone derivatives do not affect TBG levels, while progestins with androgenic activity may decrease TBG levels.[9] A combination of 30 µg/day EE and 1 mg/day norethisterone, a moderately androgenic progestin, have been found to increase TBG levels by 50 to 70%, while the combination of 30 µg/day EE and 150 µg/day desogestrel increased them by 100%.[9]
| Proteins, general | Coagulation factors | ||
|---|---|---|---|
| Compound | Effect | Compound | Effect |
| α1-Antitrypsin | + | Antithrombin III | − |
| Albumin | − | C-reactive protein | + |
| Alkaline phosphatase | + | Coagulation factor II | + |
| Angiotensinogen | + | Coagulation factor VII | + |
| Bilirubin | + | Coagulation factor VIII | + |
| Ceruloplasmin | + | Coagulation factor IX | + |
| Corticosteroid-binding globulin (transcortin) | + | Coagulation factor X | + |
| χ-Glutamyl transpeptidase | + | Coagulation factor XII | + |
| Growth hormone | + | Fibrinogen | + |
| Growth hormone-binding protein | + | Plasminogen | + |
| Insulin-like growth factor 1 | − | Protein C | + |
| Haptoglobin | − | Prothrombin time | − |
| Leucyl aminopeptidase | + | Lipids | |
| α2-Microglobulin | + | Compound | Effect |
| Orosomucoid (α1-acid glycoprotein) | − | Apolipoprotein A | + |
| Pregnancy zone protein | + | High-density lipoprotein | + |
| Retinol-binding protein | + | Low-density lipoprotein | − |
| Sex hormone-binding globulin | + | Lecithin | + |
| Thyroxine-binding globulin | + | Total lipids | + |
| Transferrin | + | Triglycerides | + |
| Key: + = Increased. − = Decreased. Sources: See template. | |||
Differences from estradiol
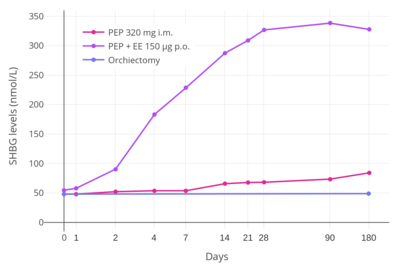
EE shows strong and disproportionate effects on liver protein synthesis relative to estradiol.[8] The liver as well as the uterus express 17β-hydroxysteroid dehydrogenase (17β-HSD), and this enzyme serves to inactivate estradiol and effectively suppress its potency in these tissues by reversibly converting it into the far less potent estrogen estrone (which has approximately 4% of the estrogenic activity of estradiol).[8] In contrast to estradiol, the 17α-ethynyl group of EE prevents oxidation of the C17β position of EE by 17β-HSD, and for this reason, EE is not inactivated in these tissues and has much stronger relative estrogenic activity in them.[8][103][11] This is the mechanism of the disproportionately strong effects of EE on hepatic protein production,[8][103] which results in a greatly increased magnitude of effect on VTE and cardiovascular risks relative to estradiol.[104]
On the other hand, due to the loss of inactivation of EE by 17β-HSD in the endometrium (uterus), EE is relatively more active than estradiol in the endometrium and, for this reason, is associated with a significantly lower incidence of vaginal bleeding and spotting in comparison.[8] This is particularly so in the case of combined estrogen and progestogen therapy (as in COCs or menopausal HRT), as progestogens induce the expression of 17β-HSD in the endometrium.[8] The reduced vaginal bleeding and spotting with EE is one of the main reasons that it is used in COCs instead of estradiol,[3] in spite of its potentially inferior safety profile (related to its adverse effects on hepatic protein synthesis and VTE incidence).[105]
EE has been found to have disproportionate effects on liver protein synthesis and VTE risk regardless of whether the route of administration is oral, transdermal, or vaginal, indicating that the use of parenteral routes over the oral route does not result in EE having proportional hepatic actions relative to non-hepatic actions.[103][9] However, the potency of EE on liver protein synthesis is in any case reduced with parenteral administration.[9] A dosage of 10 µg/day vaginal EE has been found to be equivalent to 50 µg oral EE in terms of effects on liver protein synthesis, such as stimulation of hepatic SHBG production.[9] As such, parenteral EE, which bypasses the first pass through the liver that occurs with oral EE, has been found to have a 5-fold lower impact on liver protein synthesis by weight than oral EE.[9] In contrast to EE as well as to oral estradiol, transdermal estradiol shows few or no effects on liver protein synthesis at typical menopausal dosages.[8]
| Parameters | Estradiol | Ethinylestradiol |
|---|---|---|
| ER affinity | 1 × 1010 M-1 | 2–5 × 1011 M-1 |
| Nuclear retention | 6–8 hours | 24 hours |
| Elimination half-life | 90 minutes | 7 hours |
| Substrate for 17β-HSD? | Yes | No |
| Bound to SHBG? | Yes | No |
| Relative oral liver potency | 1 | ~500–1,500 |
| Relative oral pituitary potency | 1 | 200 |
| Sources:[52] | ||
Pharmacokinetics
Absorption
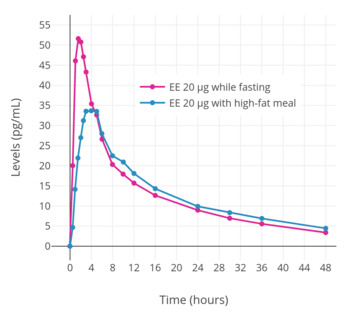
The oral bioavailability of EE is 45% on average, with a wide range of 20% to 74% (though most commonly between 38 and 48%) that is due to high interindividual variability.[10][4] Although relatively low, the oral bioavailability of EE is considerably higher than that of micronized estradiol (5%).[2][10] Following a single 20 μg dose of EE in combination with 2 mg norethisterone acetate in postmenopausal women, EE concentrations have been found to reach a maximum of 50 pg/mL within an average of 1.5 hours.[8] Following the first dose, mean levels of EE in general further increase by about 50% until steady-state concentrations are reached;[8][106] steady-state is reached after one week of daily administration.[9] For comparison, the mean peak levels of estradiol achieved with 2 mg micronized estradiol or estradiol valerate are 40 pg/mL following the first dose and 80 pg/mL after three weeks of administration.[8] These maximal concentrations of estradiol are in the same range as the concentrations of EE that are produced by an oral dose of EE that is 100 times lower by weight, which is in accordance with the approximately 100-fold increased oral potency of EE relative to estradiol.[76][8] In accordance with the high interindividual variability in the oral bioavailability of EE, there is a large degree of interindividual variation in EE levels.[8][107] A dosage of EE of 50 μg/day has been found to achieve a wide range of circulating EE levels of about 100 to 2,000 pg/mL.[108][107] Taking EE in combination with a high-fat meal has been found to significantly decrease its peak concentrations.[106][8]
EE levels after a single 50 μg dose by intravenous injection are several times higher than levels of EE after a single 50 mg dose given orally.[108] Besides the difference in levels, the course of elimination is similar for the two routes.[108]
There may be gender-specific differences in the pharmacokinetics of EE, such that EE may have greater oral potency in women than in men.[9] A study found that a combination of 60 μg/day EE and 0.25 mg/day levonorgestrel in women and men resulted in peak levels of EE of 495 pg/mL and 251 pg/mL, area-under-the-curve levels of EE of 6.216 pg/mL/hour and 2.850 pg/mL/hour, and elimination half-lives of 16.5 hours and 10.2 hours, respectively.[9] It has been suggested that this phenomenon could represent a "protection mechanism" of males against environmental estrogen exposure.[9]
Distribution
The plasma protein binding of EE is 97 to 98%, and it is bound almost exclusively to albumin.[5][8][10][109] Unlike estradiol, which binds with high affinity to SHBG, EE has very low affinity for this protein, about 2% of that of estradiol, and hence does not bind to it importantly.[110]
Metabolism
Due to high first-pass metabolism in the intestines and liver, only 1% of an oral dose of an EE appears in the circulation as EE itself.[8] During first-pass metabolism, EE is extensively conjugated via glucuronidation and sulfation into the hormonally inert ethinylestradiol glucuronides and ethinylestradiol sulfate (EE sulfate), and levels of EE sulfate in circulation are between 6- and 22-fold higher than those of EE.[8][4] For comparison, with oral administration of 2 mg micronized estradiol, levels of estrone and estrone sulfate are 4- to 6-fold and 200-fold higher than those of estradiol, respectively.[8] In contrast to estradiol, EE, due to steric hindrance by its C17α ethynyl group, is not metabolized or inactivated by 17β-HSD,[11] and this is the primary factor responsible for the dramatically increased potency of oral EE relative to oral estradiol.[8]
Aside from sulfate conjugation, EE is mainly metabolized by hydroxylation into catechol estrogens.[8] This is mainly by 2-hydroxylation into 2-hydroxy-EE, which is catalyzed primarily by CYP3A4.[10] Hydroxylation of EE at the C4, C6α, and C16β positions into 4-, 6α-, and 16β-hydroxy-EE has also been reported, but appears to contribute to its metabolism to only a small extent.[10] 2- and 4-methoxy-EE are also formed via transformation by catechol O-methyltransferase of 2- and 4-hydroxy-EE.[8] Unlike the case of estradiol, 16α-hydroxylation does not occur with EE, owing to steric hindrance by its ethynyl group at C17α.[10][8] The ethynylation of EE is largely irreversible, and so EE is not metabolized into estradiol, unlike estradiol esters.[8] A review found that the range of the reported elimination half-life of EE in the literature was 13.1 to 27.0 hours.[2] Another review reported an elimination half-life of EE of 10 to 20 hours.[10] However, the elimination half-life of EE has also been reported by other sources to be as short as 7 hours[11] and as long as 36 hours.[7]
Unlike the case of estradiol, in which there is a rapid rise in its levels and which remain elevated in a plateau-like curve for many hours, levels of EE fall rapidly after peaking.[8] This is thought to be because estrone and estrone sulfate can be reversibly converted back into estradiol and serve as a hormonally inert reservoir for estradiol, whereas the EE sulfate reservoir for EE is much smaller in comparison.[8][4] In any case, due to the formation of EE sulfate, enterohepatic recirculation is involved in the pharmacokinetics of EE similarly to estradiol, although to a lesser extent.[8][111] The contribution of enterohepatic recirculation to total circulating EE levels appears to be 12 to 20% or less, and is not observed consistently.[9][111] A secondary peak in EE levels 10 to 14 hours after administration can often be observed with oral EE.[111]
EE, following oxidative formation of a very reactive metabolite, irreversibly inhibits cytochrome P450 enzymes involved in its metabolism, and this may also play a role in the increased potency of EE relative to estradiol.[8] Indeed, EE is said to have a marked effect on hepatic metabolism, and this is one of the reasons, among others, that natural estrogens like estradiol may be preferable.[109] A 2-fold accumulation in EE levels with an EE-containing COC has been observed following 1 year of therapy.[111]
Chemistry
EE, also known as 17α-ethynylestradiol or as 17α-ethynylestra-1,3,5(10)-triene-3,17β-diol, is a synthetic estrane steroid and a derivative of estradiol with an ethynyl substitution at the C17α position.[1][112] The 17α-ethynylation of estradiol to create EE is analogous to the 17α-substitution of testosterone to make testosterone derivatives such as 17α-ethynylated progestins like ethisterone (17α-ethynyltestosterone) and norethisterone (17α-ethynyl-19-nortestosterone) as well as 17α-alkylated androgens/anabolic steroids like methyltestosterone (17α-methyltestosterone).
Analogues
A number of derivatives of EE exist.[1][112] These include mestranol (EE 3-methyl ether), quinestrol (EE 3-cyclopentyl ether), ethinylestradiol sulfonate (EE 3-isopropylsulfonate), and moxestrol (11β-methoxy-EE).[1][112][9] The former three are prodrugs of EE, while the latter one is not.[9] A few analogues of EE with other substitutions at the C17α position exist.[1][112] Examples include the estradiol derivatives methylestradiol (17α-methylestradiol) and ethylestradiol (17α-ethylestradiol), and the estriol derivatives ethinylestriol (17α-ethynylestriol) and nilestriol (17α-ethynylestriol 3-cyclopentyl ether).[1][112] Androstane analogues of EE with significant although weak estrogenic activity include ethinylandrostenediol (17α-ethynyl-5-androstenediol), 17α-ethynyl-3β-androstanediol, 17α-ethynyl-3α-androstanediol, and methandriol (17α-methyl-5-androstenediol).
History
EE was the first orally active synthetic estrogen and was described in 1938 by Hans Herloff Inhoffen and Walter Hohlweg of Schering AG in Berlin.[113][114][115][116][117] It was approved by the FDA in the U.S. on June 25, 1943 and marketed by Schering under the brand name Estinyl.[14] The FDA withdrew approval of Estinyl effective June 4, 2004 at the request of Schering, which had discontinued marketing it.[118]
EE was never introduced for use by intramuscular injection.[119]
EE was first used in COCs, as an alternative to mestranol, in 1964, and shortly thereafter superseded mestranol in COCs.[15]
Early COCs contained 40 to 100 μg/day EE and 50 to 150 μg/day mestranol.[120][121]
Society and culture
Generic names
Ethinylestradiol is the English generic name of the drug and its INN, USAN, BAN, and JAN.[122][1][123][112] It has also been spelled as ethynylestradiol, ethynyloestradiol, and ethinyloestradiol (all having the same pronunciation), and the latter was formerly its BAN but was eventually changed.[122][1][112] In addition, a space is often included in the name of EE such that it is written as ethinyl estradiol (as well as variations thereof), and this is its USP name.[122][112] The generic name of EE in French and its DCF are éthinylestradiol, in Spanish is etinilestradiol, in Italian and its DCIT are etinilestradiolo, and in Latin is ethinylestradiolum.[122][112]
The name of the drug is often abbreviated as EE or as EE2 in the medical literature.
Brand names
EE has been marketed as a standalone oral drug under the brand names Esteed, Estinyl, Feminone, Lynoral, Menolyn, Novestrol, Palonyl, Spanestrin, and Ylestrol among others, although most or all of these formulations are now discontinued.[124][125][112] It is marketed under a very large number of brand names throughout the world in combination with progestins for use as an oral contraceptive.[122] In addition, EE is marketed in the U.S. in combination with norelgestromin under the brand names Ortho Evra and Xulane as a contraceptive patch, in combination with etonogestrel under the brand name NuvaRing as a contraceptive vaginal ring, and in combination with norethisterone acetate under the brand name FemHRT in oral hormone replacement therapy for the treatment of menopausal symptoms.[12]
Availability
EE is marketed widely throughout the world.[122][112] It is marketed exclusively or almost exclusively in combination with progestins.[122]
References
- J. Elks (14 November 2014). The Dictionary of Drugs: Chemical Data: Chemical Data, Structures and Bibliographies. Springer. pp. 522–. ISBN 978-1-4757-2085-3.
- Goldzieher JW, Brody SA (1990). "Pharmacokinetics of ethinyl estradiol and mestranol". American Journal of Obstetrics and Gynecology. 163 (6 Pt 2): 2114–9. doi:10.1016/0002-9378(90)90550-Q. PMID 2256522.
- Fruzzetti F, Trémollieres F, Bitzer J (2012). "An overview of the development of combined oral contraceptives containing estradiol: focus on estradiol valerate/dienogest". Gynecological Endocrinology. 28 (5): 400–8. doi:10.3109/09513590.2012.662547. PMC 3399636. PMID 22468839.
- Fotherby K (August 1996). "Bioavailability of orally administered sex steroids used in oral contraception and hormone replacement therapy". Contraception. 54 (2): 59–69. doi:10.1016/0010-7824(96)00136-9. PMID 8842581.
- Facts and Comparisons (Firm); Ovid Technologies, Inc (2005). Drug Facts and Comparisons 2005: Pocket Version. Facts and Comparisons. p. 121. ISBN 978-1-57439-179-4.
- Micromedex (1 January 2003). USP DI 2003: Drug Information for Healthcare Professionals. Thomson Micromedex. pp. 1253, 1258, 1266. ISBN 978-1-56363-429-1.
- Claude L Hughes; Michael D. Waters (23 March 2016). Translational Toxicology: Defining a New Therapeutic Discipline. Humana Press. pp. 73–. ISBN 978-3-319-27449-2.
- Kuhl H (2005). "Pharmacology of estrogens and progestogens: influence of different routes of administration" (PDF). Climacteric. 8 Suppl 1: 3–63. doi:10.1080/13697130500148875. PMID 16112947.
- Michael Oettel; Ekkehard Schillinger (6 December 2012). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen. Springer Science & Business Media. pp. 4, 10, 15, 165, 247–248, 276–291, 363–408, 424, 514, 540, 543, 581. ISBN 978-3-642-60107-1.
The binding affinity of EE2 for the estrogen receptor is similar to that of estradiol. [...] During daily intake, the EE2 levels increase up to a steady state which is reached after about 1 week.
- Stanczyk FZ, Archer DF, Bhavnani BR (2013). "Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment". Contraception. 87 (6): 706–27. doi:10.1016/j.contraception.2012.12.011. PMID 23375353.
- Shellenberger, T. E. (1986). Pharmacology of estrogens. The Climacteric in Perspective. pp. 393–410. doi:10.1007/978-94-009-4145-8_36. ISBN 978-94-010-8339-3.
Ethinyl estradiol is a synthetic and comparatively potent estrogen. As a result of the alkylation in 17-C position it is not a substrate for 17β dehydrogenase, an enzyme which transforms natural estradiol-17β to the less potent estrone in target organs.
- "Drugs@FDA: FDA Approved Drug Products". United States Food and Drug Administration. Retrieved 22 December 2016.
- Fischer, Janos; Ganellin, C. Robin (2006). Analogue-based Drug Discovery. John Wiley & Sons. p. 482. ISBN 9783527607495.
- FDA (2007). "Approval history: Estinyl (ethinyl estradiol) NDA 005292". search: Estinyl
- J.G. Gruhn; R.R. Kazer (11 November 2013). Hormonal Regulation of the Menstrual Cycle: The Evolution of Concepts. Springer Science & Business Media. pp. 185–. ISBN 978-1-4899-3496-3.
In 1964, ethinyl estradiol was introduced as an alternative to mestranol as the estrogenic component, [...]
- Evans G, Sutton EL (2015). "Oral contraception". Med Clin North Am. 99 (3): 479–503. doi:10.1016/j.mcna.2015.01.004. PMID 25841596.
- Donna Shoupe; Florence P. Haseltine (6 December 2012). Contraception. Springer Science & Business Media. pp. 112–. ISBN 978-1-4612-2730-4.
- Hamoda, Panay, Arya, Savvas, H, N, R (2016). "The British Menopause Society & Women's Health Concern 2016 recommendations on hormone replacement therapy in menopausal women". Post Reproductive Health. 22 (4): 165–183. doi:10.1177/2053369116680501.CS1 maint: multiple names: authors list (link)
- Unger CA (2016). "Hormone therapy for transgender patients". Transl Androl Urol. 5 (6): 877–884. doi:10.21037/tau.2016.09.04. PMC 5182227. PMID 28078219.
- Coelingh Bennink HJ, Verhoeven C, Dutman AE, Thijssen J (January 2017). "The use of high-dose estrogens for the treatment of breast cancer". Maturitas. 95: 11–23. doi:10.1016/j.maturitas.2016.10.010. PMID 27889048.
- "Menopausal Hormone Therapy and Cancer Risk". American Cancer Society. February 13, 2015.
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans; World Health Organization; International Agency for Research on Cancer (2007). Combined Estrogen-progestogen Contraceptives and Combined Estrogen-progestogen Menopausal Therapy. World Health Organization. pp. 157, 433–. ISBN 978-92-832-1291-1.
- Kenneth L. Becker (2001). Principles and Practice of Endocrinology and Metabolism. Lippincott Williams & Wilkins. pp. 1024, 1027, 1035, 2153. ISBN 978-0-7817-1750-2.
Low-dose COCs contain <50 μg of estrogen and are the primary choice for oral contraception. COCs containing ≥50 μg of estrogen should no longer be routinely used for contraception. [...] The estrogen component of COCs can cause breast fullness and tenderness.
- Gregory Y. H. Lip; John E. Hall (28 June 2007). Comprehensive Hypertension E-Book. Elsevier Health Sciences. pp. 865–. ISBN 978-0-323-07067-6.
- Brian K. Alldredge; Robin L. Corelli; Michael E. Ernst (1 February 2012). Koda-Kimble and Young's Applied Therapeutics: The Clinical Use of Drugs. Lippincott Williams & Wilkins. pp. 1072–. ISBN 978-1-60913-713-7.
- "U.S. Selected Practice Recommendations for Contraceptive Use, 2016" (PDF). Recommendations and Reports. Vol. 65 no. 4. Centers for Disease Control and Prevention. July 29, 2016.
- "U.S. Selected Practice Recommendations for Contraceptive Use, 2016" (PDF). Recommendations and Reports. Vol. 65 no. 4. Centers for Disease Control and Prevention. July 29, 2016.
- "U.S. Medical Eligibility Criteria for Contraceptive Use, 2016" (PDF). Recommendations and Reports. Vol. 65 no. 3. Centers for Disease Control and Prevention. July 29, 2016.
- Jeffrey K. Aronson (21 February 2009). Meyler's Side Effects of Endocrine and Metabolic Drugs. Elsevier. pp. 177, 219, 223, 224, 230, 232, 239, 242. ISBN 978-0-08-093292-7.
- Gallo, MF; Nanda, K; Grimes, DA; Lopez, LM; Schulz, KF (1 August 2013). "20 µg versus 20 µg estrogen combined oral contraceptives for contraception". Cochrane Database of Systematic Reviews (8): CD003989. doi:10.1002/14651858.CD003989.pub5. PMC 7173696. PMID 23904209.
- Gregory Pincus (22 October 2013). Hormones and Atherosclerosis: Proceedings of the Conference Held in Brighton, Utah, March 11-14, 1958. Elsevier Science. pp. 411–. ISBN 978-1-4832-7064-7.
- Marianne J. Legato (29 October 2009). Principles of Gender-Specific Medicine. Academic Press. pp. 225–234. ISBN 978-0-08-092150-1.
- Paul D. Stein (5 April 2016). Pulmonary Embolism. Wiley. pp. 187–. ISBN 978-1-119-03909-9.
- "Combined hormonal contraception and the risk of venous thromboembolism: a guideline". Fertil. Steril. 107 (1): 43–51. January 2017. doi:10.1016/j.fertnstert.2016.09.027. PMID 27793376.
- Plu-Bureau G, Maitrot-Mantelet L, Hugon-Rodin J, Canonico M (February 2013). "Hormonal contraceptives and venous thromboembolism: an epidemiological update". Best Pract. Res. Clin. Endocrinol. Metab. 27 (1): 25–34. doi:10.1016/j.beem.2012.11.002. PMID 23384743.
- Keenan, Lynn; Kerr, Tyson; Duane, Marguerite; Van Gundy, Karl (2019). "Systematic Review of Hormonal Contraception and Risk of Venous Thrombosis". The Linacre Quarterly. 85 (4): 470–477. doi:10.1177/0024363918816683. ISSN 0024-3639. PMC 6322116.
- Tommaso Falcone; William W. Hurd (2007). Clinical Reproductive Medicine and Surgery. Elsevier Health Sciences. pp. 388–. ISBN 978-0-323-03309-1.
- Christin-Maitre S (2017). "Use of Hormone Replacement in Females with Endocrine Disorders". Horm Res Paediatr. 87 (4): 215–223. doi:10.1159/000457125. PMID 28376481.
- Gerstman BB, Gross TP, Kennedy DL, Bennett RC, Tomita DK, Stadel BV (January 1991). "Trends in the content and use of oral contraceptives in the United States, 1964-88". Am J Public Health. 81 (1): 90–6. doi:10.2105/ajph.81.1.90. PMC 1404924. PMID 1983923.
- Committee on the Relationship Between Oral Contraceptives and BreastCancer (1 January 1991). Oral Contraceptives and Breast Cancer. National Academies. pp. 143–. NAP:13774.
Following a recommendation by its Fertility and Maternal Health Drugs Advisory Committee, the Food and Drug Administration (FDA) recently ordered the removal from the market of all oral contraceptives with [ethinylestradiol] contents greater than 50 μg.
- Multigenerational Reproductive Toxicology Study of Ethinyl Estradiol (CAS No. 57636) in SpragueDawley Rats (Feed Studies). DIANE Publishing. pp. 27–. ISBN 978-1-4379-4231-6.
Oral contraceptive formulations containing greater than 50 ug ethinyl estradiol were removed from the United States market in 1989, and currently marketed formulations generally contain between 20 and 35 μg ethinyl estradiol.
- Gerstman BB, Piper JM, Tomita DK, Ferguson WJ, Stadel BV, Lundin FE (January 1991). "Oral contraceptive estrogen dose and the risk of deep venous thromboembolic disease". Am. J. Epidemiol. 133 (1): 32–7. doi:10.1093/oxfordjournals.aje.a115799. PMID 1983896.
- Sitruk-Ware R (November 2016). "Hormonal contraception and thrombosis". Fertil. Steril. 106 (6): 1289–1294. doi:10.1016/j.fertnstert.2016.08.039. PMID 27678035.
- Gallo, MF; Nanda, K; Grimes, DA; Lopez, LM; Schulz, KF (1 August 2013). "20 µg versus 20 µg estrogen combined oral contraceptives for contraception". Cochrane Database of Systematic Reviews (8): CD003989. doi:10.1002/14651858.CD003989.pub5. PMC 7173696. PMID 23904209.
- Odlind V, Milsom I, Persson I, Victor A (June 2002). "Can changes in sex hormone binding globulin predict the risk of venous thromboembolism with combined oral contraceptive pills?". Acta Obstet Gynecol Scand. 81 (6): 482–90. PMID 12047300.
- Raps M, Helmerhorst F, Fleischer K, Thomassen S, Rosendaal F, Rosing J, Ballieux B, VAN Vliet H (June 2012). "Sex hormone-binding globulin as a marker for the thrombotic risk of hormonal contraceptives". J. Thromb. Haemost. 10 (6): 992–7. doi:10.1111/j.1538-7836.2012.04720.x. PMID 22469296.
- Stanczyk FZ, Grimes DA (September 2008). "Sex hormone-binding globulin: not a surrogate marker for venous thromboembolism in women using oral contraceptives". Contraception. 78 (3): 201–3. doi:10.1016/j.contraception.2008.04.004. PMID 18692609.
- Stephen J. Winters; Ilpo T. Huhtaniemi (25 April 2017). Male Hypogonadism: Basic, Clinical and Therapeutic Principles. Humana Press. pp. 307–. ISBN 978-3-319-53298-1.
- Notelovitz M (March 2006). "Clinical opinion: the biologic and pharmacologic principles of estrogen therapy for symptomatic menopause". MedGenMed. 8 (1): 85. PMC 1682006. PMID 16915215.
- Goodman MP (February 2012). "Are all estrogens created equal? A review of oral vs. transdermal therapy". J Womens Health (Larchmt). 21 (2): 161–9. doi:10.1089/jwh.2011.2839. PMID 22011208.
- Stege R, Carlström K, Collste L, Eriksson A, Henriksson P, Pousette A (1988). "Single drug polyestradiol phosphate therapy in prostatic cancer". Am. J. Clin. Oncol. 11 Suppl 2: S101–3. doi:10.1097/00000421-198801102-00024. PMID 3242384.
- von Schoultz B, Carlström K, Collste L, Eriksson A, Henriksson P, Pousette A, Stege R (1989). "Estrogen therapy and liver function--metabolic effects of oral and parenteral administration". Prostate. 14 (4): 389–95. doi:10.1002/pros.2990140410. PMID 2664738.
- Ottosson UB, Carlström K, Johansson BG, von Schoultz B (1986). "Estrogen induction of liver proteins and high-density lipoprotein cholesterol: comparison between estradiol valerate and ethinyl estradiol". Gynecol. Obstet. Invest. 22 (4): 198–205. doi:10.1159/000298914. PMID 3817605.
- Benno Runnebaum; Thomas Rabe, eds. (17 April 2013). "Kontrazeption". Gynäkologische Endokrinologie und Fortpflanzungsmedizin: Band 1: Gynäkologische Endokrinologie. Springer-Verlag. pp. 411–512. ISBN 978-3-662-07635-4.
- Turo R, Smolski M, Esler R, Kujawa ML, Bromage SJ, Oakley N, Adeyoju A, Brown SC, Brough R, Sinclair A, Collins GN (February 2014). "Diethylstilboestrol for the treatment of prostate cancer: past, present and future" (PDF). Scand J Urol. 48 (1): 4–14. doi:10.3109/21681805.2013.861508. PMID 24256023.
- Phillips I, Shah SI, Duong T, Abel P, Langley RE (2014). "Androgen Deprivation Therapy and the Re-emergence of Parenteral Estrogen in Prostate Cancer". Oncol Hematol Rev. 10 (1): 42–47. doi:10.17925/ohr.2014.10.1.42. PMC 4052190. PMID 24932461.
- Waun Ki Hong; James F. Holland (2010). Holland-Frei Cancer Medicine 8. PMPH-USA. pp. 753–. ISBN 978-1-60795-014-1.
- Russell N, Cheung A, Grossmann M (August 2017). "Estradiol for the mitigation of adverse effects of androgen deprivation therapy". Endocr. Relat. Cancer. 24 (8): R297–R313. doi:10.1530/ERC-17-0153. PMID 28667081.
- Michael Trauner; Peter L. M. Jansen (2004). Molecular Pathogenesis of Cholestasis. Springer Science & Business Media. pp. 260–. ISBN 978-0-306-48240-3.
- Pierre-Alain Clavien; John Baillie (15 April 2008). Diseases of the Gallbladder and Bile Ducts: Diagnosis and Treatment. John Wiley & Sons. pp. 363–. ISBN 978-0-470-98697-4.
- Peter J. O'Brien; William Robert Bruce (2010). Endogenous Toxins: Diet, Genetics, Disease and Treatment. John Wiley & Sons. pp. 302–. ISBN 978-3-527-32363-0.
- A. Blaustein (11 November 2013). Pathology of the Female Genital Tract. Springer Science & Business Media. pp. 291–. ISBN 978-1-4757-1767-9.
- Earl A. Surwit; David Alberts (6 December 2012). Endometrial Cancer. Springer Science & Business Media. pp. 11–. ISBN 978-1-4613-0867-6.
- Kidd, Karen A.; Blanchfield, Paul J.; Mills, Kenneth H.; Palace, Vince P.; Evans, Robert E.; Lazorchak, James M.; Flick, Robert W. (2007-05-22). "Collapse of a fish population after exposure to a synthetic estrogen". Proceedings of the National Academy of Sciences. 104 (21): 8897–8901. doi:10.1073/pnas.0609568104. ISSN 0027-8424. PMID 17517636.
- Park, Bradley J.; Kidd, Karen (2005). "Effects of the synthetic estrogen ethinylestradiol on early life stages of mink frogs and green frogs in the wild and in situ". Environmental Toxicology and Chemistry. 24 (8): 2027–2036. doi:10.1897/04-227R.1. ISSN 1552-8618.
- Wang, Bonnie; Sanchez, Rosa I.; Franklin, Ronald B.; Evans, David C.; Huskey, Su-Er W. (November 2004). "The involvement of CYP3A4 and CYP2C9 in the metabolism of 17 alpha-ethinylestradiol". Drug Metabolism and Disposition. 32 (11): 1209–1212. doi:10.1124/dmd.104.000182. ISSN 0090-9556. PMID 15304426.
- Escande A, Pillon A, Servant N, Cravedi JP, Larrea F, Muhn P, Nicolas JC, Cavaillès V, Balaguer P (2006). "Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta". Biochem. Pharmacol. 71 (10): 1459–69. doi:10.1016/j.bcp.2006.02.002. PMID 16554039.
- Jeyakumar M, Carlson KE, Gunther JR, Katzenellenbogen JA (April 2011). "Exploration of dimensions of estrogen potency: parsing ligand binding and coactivator binding affinities". J. Biol. Chem. 286 (15): 12971–82. doi:10.1074/jbc.M110.205112. PMC 3075970. PMID 21321128.
- Prossnitz ER, Arterburn JB (July 2015). "International Union of Basic and Clinical Pharmacology. XCVII. G Protein-Coupled Estrogen Receptor and Its Pharmacologic Modulators". Pharmacol. Rev. 67 (3): 505–40. doi:10.1124/pr.114.009712. PMC 4485017. PMID 26023144.
- Yates MA, Li Y, Chlebeck PJ, Offner H (2010). "GPR30, but not estrogen receptor-alpha, is crucial in the treatment of experimental autoimmune encephalomyelitis by oral ethinyl estradiol". BMC Immunol. 11: 20. doi:10.1186/1471-2172-11-20. PMC 2864220. PMID 20403194.
- Prossnitz ER, Barton M (2011). "The G-protein-coupled estrogen receptor GPER in health and disease". Nat Rev Endocrinol. 7 (12): 715–26. doi:10.1038/nrendo.2011.122. PMC 3474542. PMID 21844907.
Further research showed that the therapeutic effect of ethynylestradiol in established EAE was mediated via GPER, but not via ERα, and possibly involved production of the anti-inflammatory cytokine Il‑10.115
- Prossnitz ER, Barton M (2014). "Estrogen biology: new insights into GPER function and clinical opportunities". Mol. Cell. Endocrinol. 389 (1–2): 71–83. doi:10.1016/j.mce.2014.02.002. PMC 4040308. PMID 24530924.
In addition, the therapeutic effect of ethinyl estradiol in established disease was demonstrated to require expression of GPER but not ERα, and was associated with the production of the anti-inflammatory cytokine IL-10 (Yates et al., 2010).
- Quaynor SD, Stradtman EW, Kim HG, Shen Y, Chorich LP, Schreihofer DA, Layman LC (July 2013). "Delayed puberty and estrogen resistance in a woman with estrogen receptor α variant". The New England Journal of Medicine. 369 (2): 164–71. doi:10.1056/NEJMoa1303611. PMC 3823379. PMID 23841731.
- Charles R. Craig; Robert E. Stitzel (2004). Modern Pharmacology with Clinical Applications. Lippincott Williams & Wilkins. pp. 708–. ISBN 978-0-7817-3762-3.
- Gautam Allahbadia; Rina Agrawal (2007). Polycystic Ovary Syndrome. Anshan. pp. 257–. ISBN 978-1-904798-74-3.
- Victor Gomel; Malcolm G. Munro; Timothy C. Rowe (1990). Gynecology: a practical approach. Williams & Wilkins. p. 132,134. ISBN 978-0-683-03631-2.
The synthetic estrogen, ethinyl estradiol, more commonly used in oral contraceptives, has a biological activity 100 times that of the native and conjugated substances.
- Donna Shoupe (7 November 2007). The Handbook of Contraception: A Guide for Practical Management. Springer Science & Business Media. pp. 23–. ISBN 978-1-59745-150-5.
EE2 has about 100 times the potency of an equivalent weight of conjugated equine estrogen or estrone sulfate for stimulating synthesis of hepatic proteins. [...] EE2 is about 1.7 times as potent as the same weight of mestranol.
- Nathaniel McConaghy (21 November 2013). Sexual Behavior: Problems and Management. Springer Science & Business Media. pp. 177–. ISBN 978-1-4899-1133-9.
Meyer et al. found that ethinyl estradiol was 75 to 100 times more potent than conjugated estrogen on the basis of the doses required to lower testosterone to the adult female range, 0.1 mg of the former and 7.5 to 10 mg of the latter being necessary.
- Bruce Chabner; Dan Louis Longo (1996). Cancer Chemotherapy and Biotherapy: Principles and Practice. Lippincott-Raven Publishers. p. 186. ISBN 978-0-397-51418-2.
The relative potency of several estrogens has been assayed by determination of effects on plasma FSH, a measure of the systemic effect, and by increases in SHBG, CBG, and angiotensinogen, all of which indicate the hepatic effect. Piperazine estrone sulfate and micronized estradiol were equipotent with respect to increases in SHBG, whereas conjugated estrogens were 3.2-fold more potent, DES was 28.4-fold more potent, and ethinyl estradiol was 600-fold more potent. With respect to decreased FSH, conjugated estrogens were 1.4-fold, DES was 3.8-fold, and ethinyl estradiol was 80 to 200-fold more potent than was piperazine estrone sulfate.
- Mashchak CA, Lobo RA, Dozono-Takano R, Eggena P, Nakamura RM, Brenner PF, Mishell DR (November 1982). "Comparison of pharmacodynamic properties of various estrogen formulations". Am. J. Obstet. Gynecol. 144 (5): 511–8. doi:10.1016/0002-9378(82)90218-6. PMID 6291391.
- Shearer RJ, Hendry WF, Sommerville IF, Fergusson JD (December 1973). "Plasma testosterone: an accurate monitor of hormone treatment in prostatic cancer". Br J Urol. 45 (6): 668–77. doi:10.1111/j.1464-410x.1973.tb12238.x. PMID 4359746.
- Ekback, Maria Palmetun (2017). "Hirsutism, What to do?" (PDF). International Journal of Endocrinology and Metabolic Disorders. 3 (3). doi:10.16966/2380-548X.140. ISSN 2380-548X.
- Eberhard Nieschlag; Hermann M. Behre; Susan Nieschlag (26 July 2012). Testosterone: Action, Deficiency, Substitution. Cambridge University Press. pp. 62–. ISBN 978-1-107-01290-5.
- Coss CC, Jones A, Parke DN, Narayanan R, Barrett CM, Kearbey JD, Veverka KA, Miller DD, Morton RA, Steiner MS, Dalton JT (2012). "Preclinical characterization of a novel diphenyl benzamide selective ERα agonist for hormone therapy in prostate cancer". Endocrinology. 153 (3): 1070–81. doi:10.1210/en.2011-1608. PMID 22294742.
- Rich P (2008). "Hormonal contraceptives for acne management". Cutis. 81 (1 Suppl): 13–8. PMID 18338653.
- Yang LP, Plosker GL (2012). "Nomegestrol acetate/estradiol: in oral contraception". Drugs. 72 (14): 1917–28. doi:10.2165/11208180-000000000-00000. PMID 22950535.
- Jacobi GH, Altwein JE, Kurth KH, Basting R, Hohenfellner R (1980). "Treatment of advanced prostatic cancer with parenteral cyproterone acetate: a phase III randomised trial". Br J Urol. 52 (3): 208–15. doi:10.1111/j.1464-410x.1980.tb02961.x. PMID 7000222.
- Gunnarsson PO, Norlén BJ (1988). "Clinical pharmacology of polyestradiol phosphate". Prostate. 13 (4): 299–304. doi:10.1002/pros.2990130405. PMID 3217277.
- Stahl F, Schnorr D, Bär CM, Fröhlich G, Dörner G (1989). "Suppression of plasma androgen levels with a combination therapy of depot-estrogen (Turisteron) and Dexamethasone in patients with prostatic cancer". Exp. Clin. Endocrinol. 94 (3): 239–43. doi:10.1055/s-0029-1210905. PMID 2630306.
- J. Larry Jameson; Leslie J. De Groot (18 May 2010). Endocrinology - E-Book: Adult and Pediatric. Elsevier Health Sciences. pp. 2282–. ISBN 978-1-4557-1126-0.
- Louis J Denis; Keith Griffiths; Amir V Kaisary; Gerald P Murphy (1 March 1999). Textbook of Prostate Cancer: Pathology, Diagnosis and Treatment: Pathology, Diagnosis and Treatment. CRC Press. pp. 297–. ISBN 978-1-85317-422-3.
- Scott WW, Menon M, Walsh PC (April 1980). "Hormonal Therapy of Prostatic Cancer". Cancer. 45 Suppl 7: 1929–1936. doi:10.1002/cncr.1980.45.s7.1929. PMID 29603164.
- Bingel AS, Benoit PS (February 1973). "Oral contraceptives: therapeutics versus adverse reactions, with an outlook for the future I". J Pharm Sci. 62 (2): 179–200. doi:10.1002/jps.2600620202. PMID 4568621.
- N. Rietbrock; A.H. Staib; D. Loew (11 March 2013). Klinische Pharmakologie: Arzneitherapie. Springer-Verlag. pp. 426–. ISBN 978-3-642-57636-2.
- Elger, Walter (1972). Physiology and pharmacology of female reproduction under the aspect of fertility control. 67. pp. 69–168. doi:10.1007/BFb0036328.
- Bastianelli, Carlo; Farris, Manuela; Rosato, Elena; Brosens, Ivo; Benagiano, Giuseppe (2018). "Pharmacodynamics of combined estrogen-progestin oral contraceptives 3. Inhibition of ovulation". Expert Review of Clinical Pharmacology. 11 (11): 1085–1098. doi:10.1080/17512433.2018.1536544. ISSN 1751-2433.
- Jorge Martinez-Manautou; Harry W. Rudel (1966). "Antiovulatory Activity of Several Synthetic and Natural Estrogens". In Robert Benjamin Greenblatt (ed.). Ovulation: Stimulation, Suppression, and Detection. Lippincott. pp. 243–253.
- Herr, F.; Revesz, C.; Manson, A. J.; Jewell, J. B. (1970). Biological Properties of Estrogen Sulfates. pp. 368–408. doi:10.1007/978-3-642-49793-3_8.
- Goldzieher JW, Pena A, Chenault CB, Woutersz TB (July 1975). "Comparative studies of the ethynyl estrogens used in oral contraceptives. II. Antiovulatory potency". Am. J. Obstet. Gynecol. 122 (5): 619–24. doi:10.1016/0002-9378(75)90061-7. PMID 1146927.
- Andrew N. Margioris; George P. Chrousos (20 April 2001). Adrenal Disorders. Springer Science & Business Media. pp. 84–. ISBN 978-1-59259-101-5.
- Polderman KH, Gooren LJ, van der Veen EA (October 1995). "Effects of gonadal androgens and oestrogens on adrenal androgen levels". Clin. Endocrinol. (Oxf). 43 (4): 415–21. doi:10.1111/j.1365-2265.1995.tb02611.x. PMID 7586614.
- Trémollieres F (February 2012). "Contraception orale estro-progestative: quelle différence entre éthinylestradiol et estradiol?" [Oral combined contraception: is there any difference between ethinyl-estradiol and estradiol?]. Gynecol Obstet Fertil (in French). 40 (2): 109–15. doi:10.1016/j.gyobfe.2011.10.009. PMID 22244780.
- Rogerio A. Lobo (5 June 2007). Treatment of the Postmenopausal Woman: Basic and Clinical Aspects. Academic Press. pp. 177, 770–771. ISBN 978-0-08-055309-2.
- Donna Shoupe (10 February 2011). Contraception. John Wiley & Sons. pp. 79–. ISBN 978-1-4443-4263-5.
- Sitruk-Ware R, Nath A (2011). "Metabolic effects of contraceptive steroids". Rev Endocr Metab Disord. 12 (2): 63–75. doi:10.1007/s11154-011-9182-4. PMID 21538049.
- Boyd RA, Zegarac EA, Eldon MA (January 2003). "The effect of food on the bioavailability of norethindrone and ethinyl estradiol from norethindrone acetate/ethinyl estradiol tablets intended for continuous hormone replacement therapy". J Clin Pharmacol. 43 (1): 52–8. doi:10.1177/0091270002239706. PMID 12520628.
- David Dodick; Stephen D. Silberstein; Stephen Silberstein (2016). Migraine. Oxford University Press. pp. 272–. ISBN 978-0-19-979361-7.
- Orme ML, Back DJ, Breckenridge AM (1983). "Clinical pharmacokinetics of oral contraceptive steroids". Clin Pharmacokinet. 8 (2): 95–136. doi:10.2165/00003088-198308020-00001. PMID 6342899.
- M. Notelovitz; P.A. van Keep (6 December 2012). The Climacteric in Perspective: Proceedings of the Fourth International Congress on the Menopause, held at Lake Buena Vista, Florida, October 28–November 2, 1984. Springer Science & Business Media. pp. 395–. ISBN 978-94-009-4145-8.
- Pugeat MM, Dunn JF, Nisula BC (July 1981). "Transport of steroid hormones: interaction of 70 drugs with testosterone-binding globulin and corticosteroid-binding globulin in human plasma". J. Clin. Endocrinol. Metab. 53 (1): 69–75. doi:10.1210/jcem-53-1-69. PMID 7195405.
- Mattison DR, Karyakina N, Goodman M, LaKind JS (September 2014). "Pharmaco- and toxicokinetics of selected exogenous and endogenous estrogens: a review of the data and identification of knowledge gaps". Crit. Rev. Toxicol. 44 (8): 696–724. doi:10.3109/10408444.2014.930813. PMID 25099693.
- Index Nominum 2000: International Drug Directory. Taylor & Francis. January 2000. p. 412. ISBN 978-3-88763-075-1.
- Inhoffen, H. H.; Hohlweg, W. (1938). "Neue per os-wirksame weibliche Keimdrüsenhormon-Derivate: 17-Aethinyl-oestradiol und Pregnen-in-on-3-ol-17 (New female glandular derivatives active per os: 17α-ethynyl-estradiol and pregnen-in-on-3-ol-17)". Naturwissenschaften. 26 (6): 96. Bibcode:1938NW.....26...96I. doi:10.1007/BF01681040.
- Maisel, Albert Q. (1965). The Hormone Quest. New York: Random House. OCLC 543168.
- Petrow, Vladimir (December 1970). "The contraceptive progestagens". Chem Rev. 70 (6): 713–26. doi:10.1021/cr60268a004. PMID 4098492.
- Sneader, Walter (2005). "Hormone analogues". Drug discovery : a history. Hoboken, NJ: John Wiley & Sons. pp. 188–225. ISBN 978-0-471-89980-8.
- Djerassi, Carl (January 2006). "Chemical birth of the pill". American Journal of Obstetrics and Gynecology. 194 (1): 290–8. doi:10.1016/j.ajog.2005.06.010. PMID 16389046.
- FDA (May 5, 2004). "Schering Corp. et al.; Withdrawal of Approval of 92 New Drug Applications and 49 Abbreviated New Drug Applications. Notice" (PDF). Federal Register. 69 (87): 25124–30.
- Cantor EB (September 1956). "A survey of estrogens". Postgrad Med. 20 (3): 224–31. doi:10.1080/00325481.1956.11691266. PMID 13359169.
- Beller, Fritz K.; Knörr, Karl; Lauritzen, Christian; Wynn, Ralph M. (1974). "Family Planning": 189–213. doi:10.1007/978-1-4615-7128-5_17. Cite journal requires
|journal=(help) - Haller, J. (1968). "Die antikonzeptionelle Therapie": 1125–1178. doi:10.1007/978-3-642-99941-3_8. Cite journal requires
|journal=(help) - "Ethinylestradiol - Drugs.com".
- I.K. Morton; Judith M. Hall (6 December 2012). Concise Dictionary of Pharmacological Agents: Properties and Synonyms. Springer Science & Business Media. pp. 115–. ISBN 978-94-011-4439-1.
- American Medical Association. Dept. of Drugs; Council on Drugs (American Medical Association); American Society for Clinical Pharmacology and Therapeutics (1 February 1977). "Estrogens, Progestagens, Oral Contraceptives, and Ovulatory Agents". AMA drug evaluations. Publishing Sciences Group. pp. 540–572. ISBN 978-0-88416-175-2.
Ethinyl Estradiol [Estinyl, Feminone, Lynoral, Novestrol, Palonyl]
- American Society of Hospital Pharmacists. Committee on Pharmacy and Pharmaceuticals (1983). American Hospital Formulary Service: A Two-volume Collection of Drug Monographs and Other Information. American Society of Hospital Pharmacists.
ETHINYL ESTRADIOL U.S.P. (Esteed®, Estinyl®, Lynoral®, Menolyn®, Novestrol®, Palonyl®, Spanestrin®, Ylestrol®)
Further reading
- Michael Oettel; Ekkehard Schillinger (6 December 2012). Estrogens and Antiestrogens II: Pharmacology and Clinical Application of Estrogens and Antiestrogen. Springer Science & Business Media. pp. 4, 10, 15, 165, 247–248, 276–291, 363–408, 424, 514, 540, 543, 581. ISBN 978-3-642-60107-1.
- Kuhl H (2005). "Pharmacology of estrogens and progestogens: influence of different routes of administration" (PDF). Climacteric. 8 Suppl 1: 3–63. doi:10.1080/13697130500148875. PMID 16112947.
- Stanczyk FZ, Archer DF, Bhavnani BR (2013). "Ethinyl estradiol and 17β-estradiol in combined oral contraceptives: pharmacokinetics, pharmacodynamics and risk assessment". Contraception. 87 (6): 706–27. doi:10.1016/j.contraception.2012.12.011. PMID 23375353.
- Mattison DR, Karyakina N, Goodman M, LaKind JS (September 2014). "Pharmaco- and toxicokinetics of selected exogenous and endogenous estrogens: a review of the data and identification of knowledge gaps". Crit. Rev. Toxicol. 44 (8): 696–724. doi:10.3109/10408444.2014.930813. PMID 25099693.