mTORC1
mTORC1, also known as mammalian target of rapamycin complex 1 or mechanistic target of rapamycin complex 1, is a protein complex that functions as a nutrient/energy/redox sensor and controls protein synthesis.[1][2]
| mTOR | |
|---|---|
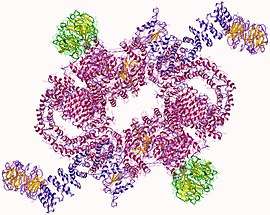 mTORC1 heteromer, Human | |
| Identifiers | |
| Symbol | MTOR |
| Alt. symbols | FRAP, FRAP2, FRAP1 |
| NCBI gene | 2475 |
| HGNC | 3942 |
| OMIM | 601231 |
| RefSeq | NM_004958 |
| UniProt | P42345 |
| Other data | |
| EC number | 2.7.11.1 |
| Locus | Chr. 1 p36 |
| RPTOR | |
|---|---|
| Identifiers | |
| Symbol | RPTOR |
| Alt. symbols | KOG1, Mip1 |
| NCBI gene | 57521 |
| HGNC | 30287 |
| OMIM | 607130 |
| RefSeq | NM_001163034.1 |
| UniProt | Q8N122 |
| Other data | |
| Locus | Chr. 17 q25.3 |
mTOR Complex 1 (mTORC1) is composed of mTOR itself, regulatory-associated protein of mTOR (Raptor), mammalian lethal with SEC13 protein 8 (MLST8), PRAS40 and DEPTOR.[2][3][4] This complex embodies the classic functions of mTOR, namely as a nutrient/energy/redox sensor and controller of protein synthesis.[1][2] The activity of this complex is regulated by rapamycin, insulin, growth factors, phosphatidic acid, certain amino acids and their derivatives (e.g., L-leucine and β-hydroxy β-methylbutyric acid), mechanical stimuli, and oxidative stress.[2][5][6]
The role of mTORC1 is to activate translation of proteins. In order for cells to grow and proliferate by manufacturing more proteins, the cells must ensure that they have the resources available for protein production. Thus, for protein production, and therefore mTORC1 activation, cells must have adequate energy resources, nutrient availability, oxygen abundance, and proper growth factors in order for mRNA translation to begin.[4]
Activation at the lysosome
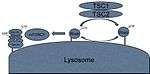
The TSC complex
Almost all of the variables required for protein synthesis affect mTORC1 activation by interacting with the TSC1/TSC2 protein complex. TSC2 is a GTPase activating protein (GAP). Its GAP activity interacts with a G protein called Rheb by hydrolyzing the GTP of the active Rheb-GTP complex, converting it to the inactive Rheb-GDP complex. The active Rheb-GTP activates mTORC1 through unelucidated pathways.[7] Thus, many of the pathways that influence mTORC1 activation do so through the activation or inactivation of the TSC1/TSC2 heterodimer. This control is usually performed through phosphorylation of the complex. This phosphorylation can cause the dimer to dissociate and lose its GAP activity, or the phosphorylation can cause the heterodimer to have increased GAP activity, depending on which amino acid residue becomes phosphorylated.[8] Thus, the signals that influence mTORC1 activity do so through activation or inactivation of the TSC1/TSC2 complex, upstream of mTORC1.
The Ragulator-Rag complex
mTORC1 interacts at the Ragulator-Rag complex on the surface of the lysosome in response to amino acid levels in the cell.[9][10] Even if a cell has the proper energy for protein synthesis, if it does not have the amino acid building blocks for proteins, no protein synthesis will occur. Studies have shown that depriving amino acid levels inhibits mTORC1 signaling to the point where both energy abundance and amino acids are necessary for mTORC1 to function. When amino acids are introduced to a deprived cell, the presence of amino acids causes Rag GTPase heterodimers to switch to their active conformation.[11] Active Rag heterodimers interact with Raptor, localizing mTORC1 to the surface of late endosomes and lysosomes where the Rheb-GTP is located.[12] This allows mTORC1 to physically interact with Rheb. Thus the amino acid pathway as well as the growth factor/energy pathway converge on endosomes and lysosomes. Thus the Ragulator-Rag complex recruits mTORC1 to lysosomes to interact with Rheb.[13][14]
Regulation of the Ragulator-Rag complex
Rag activity is regulated by at least two highly conserved complexes: the "GATOR1" complex containing DEPDC5, NPRL2 and NPRL3 and the ""GATOR2" complex containing Mios, WDR24, WDR59, Seh1L, Sec13.[15] GATOR1 inhibits Rags (it is a GTPase-activating protein for Rag subunits A/B) and GATOR2 activates Rags by inhibiting DEPDC5.
Upstream signaling
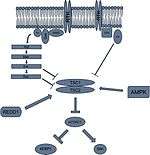
Receptor tyrosine kinases
Akt/PKB pathway
Insulin-like growth factors can activate mTORC1 through the receptor tyrosine kinase (RTK)-Akt/PKB signaling pathway. Ultimately, Akt phosphorylates TSC2 on serine residue 939, serine residue 981, and threonine residue 1462.[16] These phosphorylated sites will recruit the cytosolic anchoring protein 14-3-3 to TSC2, disrupting the TSC1/TSC2 dimer. When TSC2 is not associated with TSC1, TSC2 loses its GAP activity and can no longer hydrolyze Rheb-GTP. This results in continued activation of mTORC1, allowing for protein synthesis via insulin signaling.[17]
Akt will also phosphorylate PRAS40, causing it to fall off of the Raptor protein located on mTORC1. Since PRAS40 prevents Raptor from recruiting mTORC1's substrates 4E-BP1 and S6K1, its removal will allow the two substrates to be recruited to mTORC1 and thereby activated in this way.[18]
Furthermore, since insulin is a factor that is secreted by pancreatic beta cells upon glucose elevation in the blood, its signaling ensures that there is energy for protein synthesis to take place. In a negative feedback loop on mTORC1 signaling, S6K1 is able to phosphorylate the insulin receptor and inhibit its sensitivity to insulin.[16] This has great significance in diabetes mellitus, which is due to insulin resistance.[19]
MAPK/ERK pathway
Mitogens, such as insulin like growth factor 1 (IGF1), can activate the MAPK/ERK pathway, which can inhibit the TSC1/TSC2 complex, activating mTORC1.[17] In this pathway, the G protein Ras is tethered to the plasma membrane via a farnesyl group and is in its inactive GDP state. Upon growth factor binding to the adjacent receptor tyrosine kinase, the adaptor protein GRB2 binds with its SH2 domains. This recruits the GEF called Sos, which activates the Ras G protein. Ras activates Raf (MAPKKK), which activates Mek (MAPKK), which activates Erk (MAPK).[20] Erk can go on to activate RSK. Erk will phosphorylate the serine residue 644 on TSC2, while RSK will phosphorylate serine residue 1798 on TSC2.[21] These phosphorylations will cause the heterodimer to fall apart, and prevent it from deactivating Rheb, which keeps mTORC1 active.
RSK has also been shown to phosphorylate raptor, which helps it overcome the inhibitory effects of PRAS40.[22]
Wnt pathway
The Wnt pathway is responsible for cellular growth and proliferation during organismal development; thus, it could be reasoned that activation of this pathway also activates mTORC1. Activation of the Wnt pathway inhibits glycogen synthase kinase 3 beta (GSK3B).[23] When the Wnt pathway is not active, GSK3 beta is able to phosphorylate TSC2 on two serine residues of 1341 and 1337 in conjunction with AMPK phosphorylating serine residue 1345. It has been found that the AMPK is required to first phosphorylate residue 1345 before GSK3 beta can phosphorylate its target serine residues. This phosphorylation of TSC2 would activate this complex, if GSK3 beta were active. Since the Wnt pathway inhibits GSK3 signaling, the active Wnt pathway is also involved in the mTORC1 pathway. Thus, mTORC1 can activate protein synthesis for the developing organism.[23]
Cytokines
Cytokines like tumor necrosis factor alpha (TNF-alpha) can induce mTOR activity through IKK beta, also known as IKK2.[24] IKK beta can phosphorylate TSC1 at serine residue 487 and TSC1 at serine residue 511. This causes the heterodimer TSC complex to fall apart, keeping Rheb in its active GTP-bound state.
Energy and oxygen
Energy status
In order for translation to take place, abundant sources of energy, particularly in the form of ATP, need to be present. If these levels of ATP are not present, due to its hydrolysis into other forms like AMP, and the ratio of AMP to ATP molecules gets too high, AMPK will become activated. AMPK will go on to inhibit energy consuming pathways such as protein synthesis.[25]
AMPK can phosphorylate TSC2 on serine residue 1387, which activates the GAP activity of this complex, causing Rheb-GTP to be hydrolyzed into Rheb-GDP. This inactivates mTORC1 and blocks protein synthesis through this pathway.[26]
AMPK can also phosphorylate Raptor on two serine residues. This phosphorylated Raptor recruits 14-3-3 to bind to it and prevents Raptor from being part of the mTORC1 complex. Since mTORC1 cannot recruit its substrates without Raptor, no protein synthesis via mTORC1 occurs.[27]
LKB1, also known as STK11, is a known tumor suppressor that can activate AMPK. More studies on this aspect of mTORC1 may help shed light on its strong link to cancer.[28]
Hypoxic stress
When oxygen levels in the cell are low, it will limit its energy expenditure through the inhibition of protein synthesis. Under hypoxic conditions, hypoxia inducible factor one alpha (HIF1A) will stabilize and activate transcription of REDD1, also known as DDIT4. After translation, this REDD1 protein will bind to TSC2, which prevents 14-3-3 from inhibiting the TSC complex. Thus, TSC retains its GAP activity towards Rheb, causing Rheb to remain bound to GDP and mTORC1 to be inactive.[29][30]
Due to the lack of synthesis of ATP in the mitochondria under hypoxic stress or hypoxia, AMPK will also become active and thus inhibit mTORC1 through its processes.[31]
Downstream signaling
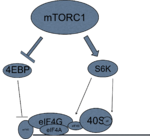
mTORC1 activates transcription and translation through its interactions with p70-S6 Kinase 1 (S6K1) and 4E-BP1, the eukaryotic initiation factor 4E (eIF4E) binding protein 1.[1] Their signaling will converge at the translation initiation complex on the 5' end of mRNA, and thus activate translation.
4E-BP1
Activated mTORC1 will phosphorylate translation inhibitor 4E-BP1, releasing it from eukaryotic translation initiation factor 4E (eIF4E).[32] eIF4E is now free to join the eukaryotic translation initiation factor 4G (eIF4G) and the eukaryotic translation initiation factor 4A (eIF4A).[33] This complex then binds to the 5' cap of mRNA and will recruit the helicase eukaryotic translation initiation factor A (eIF4A) and its cofactor eukaryotic translation initiation factor 4B (eIF4B).[34] The helicase is required to remove hairpin loops that arise in the 5' untranslated regions of mRNA, which prevent premature translation of proteins. Once the initiation complex is assembled at the 5' cap of mRNA, it will recruit the 40S small ribosomal subunit that is now capable of scanning for the AUG start codon start site, because the hairpin loop has been eradicated by the eIF4A helicase.[35] Once the ribosome reaches the AUG codon, translation can begin.
S6K
Hypophosphorylated S6K is located on the eIF3 scaffold complex. Active mTORC1 gets recruited to the scaffold, and once there, will phosphorylate S6K to make it active.[16]
mTORC1 phosphorylates S6K1 on at least two residues, with the most critical modification occurring on a threonine residue (T389).[36][37] This event stimulates the subsequent phosphorylation of S6K1 by PDPK1.[37][38] Active S6K1 can in turn stimulate the initiation of protein synthesis through activation of S6 Ribosomal protein (a component of the ribosome) and eIF4B, causing them to be recruited to the pre-initiation complex.[39]
Active S6K can bind to the SKAR scaffold protein that can get recruited to exon junction complexes (EJC). Exon junction complexes span the mRNA region where two exons come together after an intron has been spliced out. Once S6K binds to this complex, increased translation on these mRNA regions occurs.[40]
S6K1 can also participate in a positive feedback loop with mTORC1 by phosphorylating mTOR's negative regulatory domain at two sites; phosphorylation at these sites appears to stimulate mTOR activity.[41][42]
S6K also can phosphorylate programmed cell death 4 (PDCD4), which marks it for degradation by ubiquitin ligase Beta-TrCP (BTRC). PDCD4 is a tumor suppressor that binds to eIF4A and prevents it from being incorporated into the initiation complex.[43]
Role in disease and aging
mTOR was found to be related to aging in 2001 when the ortholog of S6K, SCH9, was deleted in S. cerevisiae, doubling its lifespan.[44] This greatly increased the interest in upstream signaling and mTORC1. Studies in inhibiting mTORC1 were thus performed on the model organisms of C. elegans, fruitflies, and mice. Inhibition of mTORC1 showed significantly increased lifespans in all model species.[45][46]
Based on upstream signaling of mTORC1, a clear relationship between food consumption and mTORC1 activity has been observed.[47] Most specifically, carbohydrate consumption activates mTORC1 through the insulin growth factor pathway. In addition, amino acid consumption will stimulate mTORC1 through the branched chain amino acid/Rag pathway. Thus dietary restriction inhibits mTORC1 signaling through both upstream pathways of mTORC that converge on the lysosome.[48]
Dietary restriction has been shown to significantly increase lifespan in the human model of Rhesus monkeys as well as protect against their age related decline.[49] More specifically, Rhesus monkeys on a calorie restricted diet had significantly less chance of developing cardiovascular disease, diabetes, cancer, and age related cognitive decline than those monkeys who were not placed on the calorie restricted diet.[49]
Autophagy
Autophagy is the major degradation pathway in eukaryotic cells and is essential for the removal of damaged organelles via macroautophagy or proteins and smaller cellular debris via microautophagy from the cytoplasm.[50] Thus, autophagy is a way for the cell to recycle old and damaged materials by breaking them down into their smaller components, allowing for the resynthesis of newer and healthier cellular structures.[50] Autophagy can thus remove protein aggregates and damaged organelles that can lead to cellular dysfunction.[51]
Upon activation, mTORC1 will phosphorylate autophagy-related protein 13 (Atg 13), preventing it from entering the ULK1 kinase complex, which consists of Atg1, Atg17, and Atg101.[52] This prevents the structure from being recruited to the preautophagosomal structure at the plasma membrane, inhibiting autophagy.[53]
mTORC1's ability to inhibit autophagy while at the same time stimulate protein synthesis and cell growth can result in accumulations of damaged proteins and organelles, contributing to damage at the cellular level.[54] Because autophagy appears to decline with age, activation of autophagy may help promote longevity in humans.[55] Problems in proper autophagy processes have been linked to diabetes, cardiovascular disease, neurodegenerative diseases, and cancer.[56]
Lysosomal damage
mTORC1 is positioned on lysosomes and is inhibited when lysosomal membrane is damaged through a protein complex termed GALTOR.[57] GALTOR contains galectin-8, a cytosolic lectin, which recognizes damaged lysosomal membranes by binding to the exposed glycoconjugates normally facing lysosomal lumen. Under homeostatic conditions, Galectin-8 associates with active mTOR.[57] Following membrane damage galectin-8 no longer interacts with mTOR but instead switches to complexes containing SLC38A9, RRAGA/RRAGB, and LAMTOR1 (a component of Ragulator) thus inhibiting mTOR,[57] mTOR inhibition in turn activates autophagy and starts a quality control program that removes damaged lysosomes,[57] referred to as lysophagy,[58]
Reactive oxygen species
Reactive oxygen species can damage the DNA and proteins in cells.[59] A majority of them arise in the mitochondria.[60]
Deletion of the TOR1 gene in yeast increases cellular respiration in the mitochondria by enhancing the translation of mitochondrial DNA that encodes for the complexes involved in the electron transport chain.[61] When this electron transport chain is not as efficient, the unreduced oxygen molecules in the mitochondrial cortex may accumulate and begin to produce reactive oxygen species.[62] It is important to note that both cancer cells as well as those cells with greater levels of mTORC1 both rely more on glycolysis in the cytosol for ATP production rather than through oxidative phosphorylation in the inner membrane of the mitochondria.[63]
Inhibition of mTORC1 has also been shown to increase transcription of the NFE2L2 (NRF2) gene, which is a transcription factor that is able to regulate the expression of electrophilic response elements as well as antioxidants in response to increased levels of reactive oxygen species.[64]
Though AMPK induced eNOS has been shown to regulate mTORC1 in endothelium. Unlike the other cell type in endothelium eNOS induced mTORC1 and this pathway is required for mitochondrial biogenesis.[65]
Stem cells
Conservation of stem cells in the body has been shown to help prevent against premature aging.[66] mTORC1 activity plays a critical role in the growth and proliferation of stem cells.[67] Knocking out mTORC1 results in embryonic lethality due to lack of trophoblast development.[68] Treating stem cells with rapamycin will also slow their proliferation, conserving the stem cells in their undifferentiated condition.[67]
mTORC1 plays a role in the differentiation and proliferation of hematopoietic stem cells. Its upregulation has been shown to cause premature aging in hematopoietic stem cells. Conversely, inhibiting mTOR restores and regenerates the hematopoietic stem cell line.[69] The mechanisms of mTORC1's inhibition on proliferation and differentiation of hematopoietic stem cells has yet to be fully elucidated.[70]
Rapamycin is used clinically as an immunosuppressant and prevents the proliferation of T cells and B cells.[71] Paradoxically, even though rapamycin is a federally approved immunosuppressant, its inhibition of mTORC1 results in better quantity and quality of functional memory T cells. mTORC1 inhibition with rapamycin improves the ability of naïve T cells to become precursor memory T cells during the expansion phase of T cell development .[72] This inhibition also allows for an increase in quality of these memory T cells that become mature T cells during the contraction phase of their development.[73] mTORC1 inhibition with rapamycin has also been linked to a dramatic increase of B cells in old mice, enhancing their immune systems.[69] This paradox of rapamycin inhibiting the immune system response has been linked to several reasons, including its interaction with regulatory T cells.[73]
As a biomolecular target
Activators
Resistance exercise, the amino acid L-leucine, and beta-hydroxy beta-methylbutyric acid (HMB) are known to induce signaling cascades in skeletal muscle cells that result in mTOR phosphorylation, the activation of mTORC1, and subsequently the initiation of myofibrillar protein synthesis (i.e., the production of proteins such as myosin, titin, and actin), thereby facilitating muscle hypertrophy.
The NMDA receptor antagonist ketamine has been found to activate the mTORC1 pathway in the medial prefrontal cortex (mPFC) of the brain as an essential downstream mechanism in the mediation of its rapid-acting antidepressant effects.[74] NV-5138 is a ligand and modulator of sestrin2, a leucine amino acid sensor and upstream regulatory pathway of mTORC1, and is under development for the treatment of depression.[74] The drug has been found to directly and selectively activate the mTORC1 pathway, including in the mPFC, and to produce rapid-acting antidepressant effects similar to those of ketamine.[74]
Inhibitors
There have been several dietary compounds that have been suggested to inhibit mTORC1 signaling including EGCG, resveratrol, curcumin, caffeine, and alcohol.[75][76]
First generation drugs
Rapamycin was the first known inhibitor of mTORC1, considering that mTORC1 was discovered as being the target of rapamycin.[77] Rapamycin will bind to cytosolic FKBP12 and act as a scaffold molecule, allowing this protein to dock on the FRB regulatory region (FKBP12-Rapamycin Binding region/domain) on mTORC1.[78] The binding of the FKBP12-rapamycin complex to the FRB regulatory region inhibits mTORC1 through processes not yet known. mTORC2 is also inhibited by rapamycin in some cell culture lines and tissues, particularly those that express high levels of FKBP12 and low levels of FKBP51.[79][80][81]
Rapamycin itself is not very water soluble and is not very stable, so scientists developed rapamycin analogs, called rapalogs, to overcome these two problems with rapamycin.[82] These drugs are considered the first generation inhibitors of mTOR.[83] These other inhibitors include everolimus and temsirolimus.
Sirolimus, which is the drug name for rapamycin, was approved by the U.S. Food and Drug Administration (FDA) in 1999 to prevent against transplant rejection in patients undergoing kidney transplantation.[84] In 2003, it was approved as a stent covering for widening arteries to prevent against future heart attacks.[85] In 2007, mTORC1 inhibitors began being approved for treatments against cancers such as renal cell carcinoma.[86] In 2008 they were approved for treatment of mantle cell lymphoma.[87] mTORC1 inhibitors have recently been approved for treatment of pancreatic cancer.[88] In 2010 they were approved for treatment of tuberous sclerosis.[89]
Second generation drugs
The second generation of inhibitors were created to overcome problems with upstream signaling upon the introduction of first generation inhibitors to the treated cells.[90] One problem with the first generation inhibitors of mTORC1 is that there is a negative feedback loop from phosphorylated S6K, that can inhibit the insulin RTK via phosphorylation.[91] When this negative feedback loop is no longer there, the upstream regulators of mTORC1 become more active than they would otherwise would have been under normal mTORC1 activity. Another problem is that since mTORC2 is resistant to rapamycin, and it too acts upstream of mTORC1 by activating Akt.[82] Thus signaling upstream of mTORC1 still remains very active upon its inhibition via rapamycin and the rapalogs.
Second generation inhibitors are able to bind to the ATP-binding motif on the kinase domain of the mTOR core protein itself and abolish activity of both mTOR complexes.[90] In addition, since the mTOR and the PI3K proteins are both in the same phosphatidylinositol 3-kinase-related kinase (PIKK) family of kinases, some second generation inhibitors have dual inhibition towards the mTOR complexes as well as PI3K, which acts upstream of mTORC1.[82] As of 2011, these second generation inhibitors were in phase II of clinical trials.
Third generation drugs
The third generation of inhibitors were created following the realization that many of the side effects of rapamycin and rapamycin analogs were mediated not as a result of direct inhibition of mTORC1, but as a consequence of off-target inhibition of mTORC2.[92][93] Rapamycin analogs that are more selective for mTORC1 than sirolimus have been developed and in mice have reduced side effects.[94] mTORC1 inhibitors that have novel mechanisms of action - for example, inhibiting the interaction of mTORC1 with its activator Rheb - are also being developed.[95]
There have been over 1,300 clinical trials conducted with mTOR inhibitors since 1970.[96]
References
- Hay N, Sonenberg N (Aug 2004). "Upstream and downstream of mTOR". Genes & Development. 18 (16): 1926–45. doi:10.1101/gad.1212704. PMID 15314020.
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM (Jul 2002). "mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery". Cell. 110 (2): 163–75. doi:10.1016/S0092-8674(02)00808-5. PMID 12150925.
- Kim DH, Sarbassov DD, Ali SM, Latek RR, Guntur KV, Erdjument-Bromage H, Tempst P, Sabatini DM (Apr 2003). "GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR". Molecular Cell. 11 (4): 895–904. doi:10.1016/S1097-2765(03)00114-X. PMID 12718876.
- Wullschleger S, Loewith R, Hall MN (Feb 2006). "TOR signaling in growth and metabolism". Cell. 124 (3): 471–84. doi:10.1016/j.cell.2006.01.016. PMID 16469695.
- Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J (Nov 2001). "Phosphatidic acid-mediated mitogenic activation of mTOR signaling". Science. 294 (5548): 1942–5. Bibcode:2001Sci...294.1942F. doi:10.1126/science.1066015. PMID 11729323.
- Bond P (March 2016). "Regulation of mTORC1 by growth factors, energy status, amino acids and mechanical stimuli at a glance". J. Int. Soc. Sports Nutr. 13: 8. doi:10.1186/s12970-016-0118-y. PMC 4774173. PMID 26937223.
- Beauchamp EM, Platanias LC (Aug 2013). "The evolution of the TOR pathway and its role in cancer". Oncogene. 32 (34): 3923–32. doi:10.1038/onc.2012.567. PMID 23246968.
- Durán RV, Hall MN (Feb 2012). "Regulation of TOR by small GTPases". EMBO Reports. 13 (2): 121–8. doi:10.1038/embor.2011.257. PMC 3271343. PMID 22240970.
- Jewell JL, Russell RC, Guan KL (Mar 2013). "Amino acid signalling upstream of mTOR". Nature Reviews Molecular Cell Biology. 14 (3): 133–9. doi:10.1038/nrm3522. PMC 3988467. PMID 23361334.
- Efeyan, Alejo; Zoncu, Roberto; Sabatini, David M. (2012-09-01). "Amino acids and mTORC1: from lysosomes to disease". Trends in Molecular Medicine. 18 (9): 524–533. doi:10.1016/j.molmed.2012.05.007. hdl:1721.1/106904. ISSN 1471-4914. PMC 3432651. PMID 22749019.
- Efeyan A, Zoncu R, Sabatini DM (Sep 2012). "Amino acids and mTORC1: from lysosomes to disease". Trends in Molecular Medicine. 18 (9): 524–33. doi:10.1016/j.molmed.2012.05.007. PMC 3432651. PMID 22749019.
- Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM (Jun 2008). "The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1". Science. 320 (5882): 1496–501. Bibcode:2008Sci...320.1496S. doi:10.1126/science.1157535. PMC 2475333. PMID 18497260.
- Saucedo LJ, Gao X, Chiarelli DA, Li L, Pan D, Edgar BA (Jun 2003). "Rheb promotes cell growth as a component of the insulin/TOR signalling network". Nature Cell Biology. 5 (6): 566–71. doi:10.1038/ncb996. PMID 12766776.
- Suzuki T, Inoki K (Sep 2011). "Spatial regulation of the mTORC1 system in amino acids sensing pathway". Acta Biochimica et Biophysica Sinica. 43 (9): 671–9. doi:10.1093/abbs/gmr066. PMC 3160786. PMID 21785113.
- Bar-Peled L, Chantranupong L, Cherniack AD, Chen WW, Ottina KA, Grabiner BC, Spear ED, Carter SL, Meyerson M, Sabatini DM (May 2013). "A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1". Science. 340 (6136): 1100–6. Bibcode:2013Sci...340.1100B. doi:10.1126/science.1232044. PMC 3728654. PMID 23723238.
- Ma XM, Blenis J (May 2009). "Molecular mechanisms of mTOR-mediated translational control". Nature Reviews Molecular Cell Biology. 10 (5): 307–18. doi:10.1038/nrm2672. PMID 19339977.
- Mendoza MC, Er EE, Blenis J (Jun 2011). "The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation". Trends in Biochemical Sciences. 36 (6): 320–8. doi:10.1016/j.tibs.2011.03.006. PMC 3112285. PMID 21531565.
- Oshiro N, Takahashi R, Yoshino K, Tanimura K, Nakashima A, Eguchi S, Miyamoto T, Hara K, Takehana K, Avruch J, Kikkawa U, Yonezawa K (Jul 2007). "The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1". The Journal of Biological Chemistry. 282 (28): 20329–39. doi:10.1074/jbc.M702636200. PMC 3199301. PMID 17517883.
- Ye J (Mar 2013). "Mechanisms of insulin resistance in obesity". Frontiers of Medicine. 7 (1): 14–24. doi:10.1007/s11684-013-0262-6. PMC 3936017. PMID 23471659.
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S, Malaponte G, Mazzarino MC, Fagone P, Nicoletti F, Bäsecke J, Mijatovic S, Maksimovic-Ivanic D, Milella M, Tafuri A, Chiarini F, Evangelisti C, Cocco L, Martelli AM (Oct 2012). "Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance". Oncotarget. 3 (10): 1068–111. doi:10.18632/oncotarget.659. PMC 3717945. PMID 23085539.
- Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP (Apr 2005). "Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis". Cell. 121 (2): 179–93. doi:10.1016/j.cell.2005.02.031. PMID 15851026.
- Carrière A, Cargnello M, Julien LA, Gao H, Bonneil E, Thibault P, Roux PP (Sep 2008). "Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation". Current Biology. 18 (17): 1269–77. doi:10.1016/j.cub.2008.07.078. PMID 18722121.
- Majid S, Saini S, Dahiya R (2012). "Wnt signaling pathways in urological cancers: past decades and still growing". Molecular Cancer. 11: 7. doi:10.1186/1476-4598-11-7. PMC 3293036. PMID 22325146.
- Salminen A, Hyttinen JM, Kauppinen A, Kaarniranta K (2012). "Context-Dependent Regulation of Autophagy by IKK-NF-κB Signaling: Impact on the Aging Process". International Journal of Cell Biology. 2012: 849541. doi:10.1155/2012/849541. PMC 3412117. PMID 22899934.
- Hardie DG (Oct 2007). "AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy". Nature Reviews Molecular Cell Biology. 8 (10): 774–85. doi:10.1038/nrm2249. PMID 17712357.
- Mihaylova MM, Shaw RJ (Sep 2011). "The AMPK signalling pathway coordinates cell growth, autophagy and metabolism". Nature Cell Biology. 13 (9): 1016–23. doi:10.1038/ncb2329. PMC 3249400. PMID 21892142.
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, Shaw RJ (Apr 2008). "AMPK phosphorylation of raptor mediates a metabolic checkpoint". Molecular Cell. 30 (2): 214–26. doi:10.1016/j.molcel.2008.03.003. PMC 2674027. PMID 18439900.
- Nagalingam A, Arbiser JL, Bonner MY, Saxena NK, Sharma D (2012). "Honokiol activates AMP-activated protein kinase in breast cancer cells via an LKB1-dependent pathway and inhibits breast carcinogenesis". Breast Cancer Research. 14 (1): R35. doi:10.1186/bcr3128. PMC 3496153. PMID 22353783.
- Horak P, Crawford AR, Vadysirisack DD, Nash ZM, DeYoung MP, Sgroi D, Ellisen LW (Mar 2010). "Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis". Proceedings of the National Academy of Sciences of the United States of America. 107 (10): 4675–80. Bibcode:2010PNAS..107.4675H. doi:10.1073/pnas.0907705107. PMC 2842042. PMID 20176937.
- Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG (Dec 2004). "Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex". Genes & Development. 18 (23): 2893–904. doi:10.1101/gad.1256804. PMC 534650. PMID 15545625.
- Wang S, Song P, Zou MH (Jun 2012). "AMP-activated protein kinase, stress responses and cardiovascular diseases". Clinical Science. 122 (12): 555–73. doi:10.1042/CS20110625. PMC 3367961. PMID 22390198.
- Martelli AM, Evangelisti C, Chappell W, Abrams SL, Bäsecke J, Stivala F, Donia M, Fagone P, Nicoletti F, Libra M, Ruvolo V, Ruvolo P, Kempf CR, Steelman LS, McCubrey JA (Jul 2011). "Targeting the translational apparatus to improve leukemia therapy: roles of the PI3K/PTEN/Akt/mTOR pathway". Leukemia. 25 (7): 1064–79. doi:10.1038/leu.2011.46. PMID 21436840.
- Wang H, Zhang Q, Wen Q, Zheng Y, Lazarovici P, Philip L, Jiang H, Lin J, Zheng W (Jan 2012). "Proline-rich Akt substrate of 40kDa (PRAS40): a novel downstream target of PI3k/Akt signaling pathway". Cellular Signalling. 24 (1): 17–24. doi:10.1016/j.cellsig.2011.08.010. PMID 21906675.
- Raught B, Gingras AC (Jan 1999). "eIF4E activity is regulated at multiple levels". The International Journal of Biochemistry & Cell Biology. 31 (1): 43–57. doi:10.1016/s1357-2725(98)00131-9. PMID 10216943.
- Lee T, Pelletier J (Jan 2012). "Eukaryotic initiation factor 4F: a vulnerability of tumor cells". Future Medicinal Chemistry. 4 (1): 19–31. doi:10.4155/fmc.11.150. PMID 22168162.
- Saitoh M, Pullen N, Brennan P, Cantrell D, Dennis PB, Thomas G (May 2002). "Regulation of an activated S6 kinase 1 variant reveals a novel mammalian target of rapamycin phosphorylation site". The Journal of Biological Chemistry. 277 (22): 20104–12. doi:10.1074/jbc.M201745200. PMID 11914378.
- Pullen N, Thomas G (Jun 1997). "The modular phosphorylation and activation of p70s6k". FEBS Letters. 410 (1): 78–82. doi:10.1016/S0014-5793(97)00323-2. PMID 9247127.
- Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas G (Jan 1998). "Phosphorylation and activation of p70s6k by PDK1". Science. 279 (5351): 707–10. Bibcode:1998Sci...279..707P. doi:10.1126/science.279.5351.707. PMID 9445476.
- Peterson RT, Schreiber SL (Mar 1998). "Translation control: connecting mitogens and the ribosome". Current Biology. 8 (7): R248–50. doi:10.1016/S0960-9822(98)70152-6. PMID 9545190.
- Ma XM, Yoon SO, Richardson CJ, Jülich K, Blenis J (Apr 2008). "SKAR links pre-mRNA splicing to mTOR/S6K1-mediated enhanced translation efficiency of spliced mRNAs". Cell. 133 (2): 303–13. doi:10.1016/j.cell.2008.02.031. PMID 18423201.
- Chiang GG, Abraham RT (Jul 2005). "Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase". The Journal of Biological Chemistry. 280 (27): 25485–90. doi:10.1074/jbc.M501707200. PMID 15899889.
- Holz MK, Blenis J (Jul 2005). "Identification of S6 kinase 1 as a novel mammalian target of rapamycin (mTOR)-phosphorylating kinase". The Journal of Biological Chemistry. 280 (28): 26089–93. doi:10.1074/jbc.M504045200. PMID 15905173.
- Schmid T, Jansen AP, Baker AR, Hegamyer G, Hagan JP, Colburn NH (Mar 2008). "Translation inhibitor Pdcd4 is targeted for degradation during tumor promotion". Cancer Research. 68 (5): 1254–60. doi:10.1158/0008-5472.CAN-07-1719. PMID 18296647.
- Fabrizio P, Pozza F, Pletcher SD, Gendron CM, Longo VD (Apr 2001). "Regulation of longevity and stress resistance by Sch9 in yeast". Science. 292 (5515): 288–90. Bibcode:2001Sci...292..288F. doi:10.1126/science.1059497. PMID 11292860.
- Robida-Stubbs S, Glover-Cutter K, Lamming DW, Mizunuma M, Narasimhan SD, Neumann-Haefelin E, Sabatini DM, Blackwell TK (May 2012). "TOR signaling and rapamycin influence longevity by regulating SKN-1/Nrf and DAF-16/FoxO". Cell Metabolism. 15 (5): 713–24. doi:10.1016/j.cmet.2012.04.007. PMC 3348514. PMID 22560223.
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (Jul 2009). "Rapamycin fed late in life extends lifespan in genetically heterogeneous mice". Nature. 460 (7253): 392–5. Bibcode:2009Natur.460..392H. doi:10.1038/nature08221. PMC 2786175. PMID 19587680.
- Kaeberlein M, Powers RW, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK (Nov 2005). "Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients". Science. 310 (5751): 1193–6. Bibcode:2005Sci...310.1193K. doi:10.1126/science.1115535. PMID 16293764.
- Blagosklonny MV (Feb 2010). "Calorie restriction: decelerating mTOR-driven aging from cells to organisms (including humans)". Cell Cycle. 9 (4): 683–8. doi:10.4161/cc.9.4.10766. PMID 20139716.
- Colman RJ, Anderson RM, Johnson SC, Kastman EK, Kosmatka KJ, Beasley TM, Allison DB, Cruzen C, Simmons HA, Kemnitz JW, Weindruch R (Jul 2009). "Caloric restriction delays disease onset and mortality in rhesus monkeys". Science. 325 (5937): 201–4. Bibcode:2009Sci...325..201C. doi:10.1126/science.1173635. PMC 2812811. PMID 19590001.
- Choi AM, Ryter SW, Levine B (Feb 2013). "Autophagy in human health and disease". The New England Journal of Medicine. 368 (7): 651–62. doi:10.1056/NEJMra1205406. PMID 23406030.
- Murrow L, Debnath J (Jan 2013). "Autophagy as a stress-response and quality-control mechanism: implications for cell injury and human disease". Annual Review of Pathology. 8: 105–37. doi:10.1146/annurev-pathol-020712-163918. PMC 3971121. PMID 23072311.
- Alers S, Löffler AS, Wesselborg S, Stork B (Jan 2012). "Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks". Molecular and Cellular Biology. 32 (1): 2–11. doi:10.1128/MCB.06159-11. PMC 3255710. PMID 22025673.
- Pyo JO, Nah J, Jung YK (Feb 2012). "Molecules and their functions in autophagy". Experimental & Molecular Medicine. 44 (2): 73–80. doi:10.3858/emm.2012.44.2.029. PMC 3296815. PMID 22257882.
- Proud CG (Nov 2007). "Amino acids and mTOR signalling in anabolic function". Biochemical Society Transactions. 35 (Pt 5): 1187–90. doi:10.1042/BST0351187. PMID 17956308.
- Cuervo AM, Dice JF (Oct 2000). "Age-related decline in chaperone-mediated autophagy". The Journal of Biological Chemistry. 275 (40): 31505–13. doi:10.1074/jbc.M002102200. PMID 10806201.
- Codogno P, Meijer AJ (Nov 2005). "Autophagy and signaling: their role in cell survival and cell death". Cell Death and Differentiation. 12 Suppl 2: 1509–18. doi:10.1038/sj.cdd.4401751. PMID 16247498.
- Jia J, Abudu YP, Claude-Taupin A, Gu Y, Kumar S, Choi SW, Peters R, Mudd MH, Allers L, Salemi M, Phinney B, Johansen T, Deretic V (April 2018). "Galectins Control mTOR in Response to Endomembrane Damage". Molecular Cell. 70 (1): 120–135.e8. doi:10.1016/j.molcel.2018.03.009. PMC 5911935. PMID 29625033.
- Hasegawa J, Maejima I, Iwamoto R, Yoshimori T (March 2015). "Selective autophagy: lysophagy". Methods. 75: 128–32. doi:10.1016/j.ymeth.2014.12.014. PMID 25542097.
- Apel K, Hirt H (2004). "Reactive oxygen species: metabolism, oxidative stress, and signal transduction". Annual Review of Plant Biology. 55: 373–99. doi:10.1146/annurev.arplant.55.031903.141701. PMID 15377225.
- Murphy MP (Jan 2009). "How mitochondria produce reactive oxygen species". The Biochemical Journal. 417 (1): 1–13. doi:10.1042/BJ20081386. PMC 2605959. PMID 19061483.
- Bonawitz ND, Chatenay-Lapointe M, Pan Y, Shadel GS (Apr 2007). "Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression". Cell Metabolism. 5 (4): 265–77. doi:10.1016/j.cmet.2007.02.009. PMC 3460550. PMID 17403371.
- Adam-Vizi V (2005). "Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources". Antioxidants & Redox Signaling. 7 (9–10): 1140–9. doi:10.1089/ars.2005.7.1140. PMID 16115017.
- Sun Q, Chen X, Ma J, Peng H, Wang F, Zha X, Wang Y, Jing Y, Yang H, Chen R, Chang L, Zhang Y, Goto J, Onda H, Chen T, Wang MR, Lu Y, You H, Kwiatkowski D, Zhang H (Mar 2011). "Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth". Proceedings of the National Academy of Sciences of the United States of America. 108 (10): 4129–34. Bibcode:2011PNAS..108.4129S. doi:10.1073/pnas.1014769108. PMC 3054028. PMID 21325052.
- Sporn MB, Liby KT (Aug 2012). "NRF2 and cancer: the good, the bad and the importance of context". Nature Reviews. Cancer. 12 (8): 564–71. doi:10.1038/nrc3278. PMC 3836441. PMID 22810811.
- Li C, Reif MM, Craige S, Kant S, Keaney JF (Mar 2016). "Endothelial AMPK Activation Induces Mitochondrial Biogenesis and Stress Adaptation via eNOS-Dependent mTORC1 Signalingt". Nitric Oxide. 55–56: 45–53. doi:10.1016/j.niox.2016.03.003. PMC 4860108. PMID 26989010.
- Ho AD, Wagner W, Mahlknecht U (Jul 2005). "Stem cells and ageing. The potential of stem cells to overcome age-related deteriorations of the body in regenerative medicine". EMBO Reports. 6 Spec No: S35–8. doi:10.1038/sj.embor.7400436. PMC 1369281. PMID 15995659.
- Murakami M, Ichisaka T, Maeda M, Oshiro N, Hara K, Edenhofer F, Kiyama H, Yonezawa K, Yamanaka S (Aug 2004). "mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells". Molecular and Cellular Biology. 24 (15): 6710–8. doi:10.1128/MCB.24.15.6710-6718.2004. PMC 444840. PMID 15254238.
- Gangloff YG, Mueller M, Dann SG, Svoboda P, Sticker M, Spetz JF, Um SH, Brown EJ, Cereghini S, Thomas G, Kozma SC (Nov 2004). "Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development". Molecular and Cellular Biology. 24 (21): 9508–16. doi:10.1128/MCB.24.21.9508-9516.2004. PMC 522282. PMID 15485918.
- Chen C, Liu Y, Liu Y, Zheng P (2009). "mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells". Science Signaling. 2 (98): ra75. doi:10.1126/scisignal.2000559. PMC 4020596. PMID 19934433.
- Russell RC, Fang C, Guan KL (Aug 2011). "An emerging role for TOR signaling in mammalian tissue and stem cell physiology". Development. 138 (16): 3343–56. doi:10.1242/dev.058230. PMC 3143559. PMID 21791526.
- Limon JJ, Fruman DA (2012). "Akt and mTOR in B Cell Activation and Differentiation". Frontiers in Immunology. 3: 228. doi:10.3389/fimmu.2012.00228. PMC 3412259. PMID 22888331.
- Araki K, Turner AP, Shaffer VO, Gangappa S, Keller SA, Bachmann MF, Larsen CP, Ahmed R (Jul 2009). "mTOR regulates memory CD8 T-cell differentiation". Nature. 460 (7251): 108–12. Bibcode:2009Natur.460..108A. doi:10.1038/nature08155. PMC 2710807. PMID 19543266.
- Araki K, Youngblood B, Ahmed R (May 2010). "The role of mTOR in memory CD8 T-cell differentiation". Immunological Reviews. 235 (1): 234–43. doi:10.1111/j.0105-2896.2010.00898.x. PMC 3760155. PMID 20536567.
- Duman RS (2018). "Ketamine and rapid-acting antidepressants: a new era in the battle against depression and suicide". F1000Res. 7: 659. doi:10.12688/f1000research.14344.1. PMC 5968361. PMID 29899972.
- Liu M, Wilk SA, Wang A, Zhou L, Wang RH, Ogawa W, Deng C, Dong LQ, Liu F (Nov 2010). "Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and DEPTOR". The Journal of Biological Chemistry. 285 (47): 36387–94. doi:10.1074/jbc.M110.169284. PMC 2978567. PMID 20851890.
- Miwa S, Sugimoto N, Yamamoto N, Shirai T, Nishida H, Hayashi K, Kimura H, Takeuchi A, Igarashi K, Yachie A, Tsuchiya H (Sep 2012). "Caffeine induces apoptosis of osteosarcoma cells by inhibiting AKT/mTOR/S6K, NF-κB and MAPK pathways". Anticancer Research. 32 (9): 3643–9. PMID 22993301.
- Vézina C, Kudelski A, Sehgal SN (October 1975). "Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle". The Journal of Antibiotics. 28 (10): 721–6. doi:10.7164/antibiotics.28.721. PMID 1102508.
- Tsang CK, Qi H, Liu LF, Zheng XF (February 2007). "Targeting mammalian target of rapamycin (mTOR) for health and diseases". Drug Discovery Today. 12 (3–4): 112–24. doi:10.1016/j.drudis.2006.12.008. PMID 17275731.
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM (April 2006). "Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB". Molecular Cell. 22 (2): 159–68. doi:10.1016/j.molcel.2006.03.029. PMID 16603397.
- Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM, Baur JA (March 2012). "Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity". Science. 335 (6076): 1638–43. Bibcode:2012Sci...335.1638L. doi:10.1126/science.1215135. PMC 3324089. PMID 22461615.
- Schreiber KH, Ortiz D, Academia EC, Anies AC, Liao CY, Kennedy BK (April 2015). "Rapamycin-mediated mTORC2 inhibition is determined by the relative expression of FK506-binding proteins". Aging Cell. 14 (2): 265–73. doi:10.1111/acel.12313. PMC 4364838. PMID 25652038.
- Vilar E, Perez-Garcia J, Tabernero J (Mar 2011). "Pushing the envelope in the mTOR pathway: the second generation of inhibitors". Molecular Cancer Therapeutics. 10 (3): 395–403. doi:10.1158/1535-7163.MCT-10-0905. PMC 3413411. PMID 21216931.
- De P, Miskimins K, Dey N, Leyland-Jones B (Aug 2013). "Promise of rapalogues versus mTOR kinase inhibitors in subset specific breast cancer: old targets new hope". Cancer Treatment Reviews. 39 (5): 403–12. doi:10.1016/j.ctrv.2012.12.002. PMID 23352077.
- Nashan B, Citterio F (Sep 2012). "Wound healing complications and the use of mammalian target of rapamycin inhibitors in kidney transplantation: a critical review of the literature". Transplantation. 94 (6): 547–61. doi:10.1097/TP.0b013e3182551021. PMID 22941182.
- Townsend JC, Rideout P, Steinberg DH (2012). "Everolimus-eluting stents in interventional cardiology". Vascular Health and Risk Management. 8: 393–404. doi:10.2147/VHRM.S23388. PMC 3402052. PMID 22910420.
- Voss MH, Molina AM, Motzer RJ (Aug 2011). "mTOR inhibitors in advanced renal cell carcinoma". Hematology/Oncology Clinics of North America. 25 (4): 835–52. doi:10.1016/j.hoc.2011.04.008. PMC 3587783. PMID 21763970.
- Smith SM (Jun 2012). "Targeting mTOR in mantle cell lymphoma: current and future directions". Best Practice & Research. Clinical Haematology. 25 (2): 175–83. doi:10.1016/j.beha.2012.04.008. PMID 22687453.
- Fasolo A, Sessa C (2012). "Targeting mTOR pathways in human malignancies". Current Pharmaceutical Design. 18 (19): 2766–77. doi:10.2174/138161212800626210. PMID 22475451.
- Budde K, Gaedeke J (Feb 2012). "Tuberous sclerosis complex-associated angiomyolipomas: focus on mTOR inhibition". American Journal of Kidney Diseases. 59 (2): 276–83. doi:10.1053/j.ajkd.2011.10.013. PMID 22130643.
- Zhang YJ, Duan Y, Zheng XF (Apr 2011). "Targeting the mTOR kinase domain: the second generation of mTOR inhibitors". Drug Discovery Today. 16 (7–8): 325–31. doi:10.1016/j.drudis.2011.02.008. PMC 3073023. PMID 21333749.
- Veilleux A, Houde VP, Bellmann K, Marette A (Apr 2010). "Chronic inhibition of the mTORC1/S6K1 pathway increases insulin-induced PI3K activity but inhibits Akt2 and glucose transport stimulation in 3T3-L1 adipocytes". Molecular Endocrinology (Baltimore, Md.). 24 (4): 766–78. doi:10.1210/me.2009-0328. PMC 5417537. PMID 20203102.
- Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, et al. (March 2012). "Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity". Science. 335 (6076): 1638–43. Bibcode:2012Sci...335.1638L. doi:10.1126/science.1215135. PMC 3324089. PMID 22461615.
- Zhou H, Huang S (2016). "Role of mTOR signaling in tumor cell motility, invasion and metastasis". In Atta-ur-Rahman (ed.). Advances in Cancer Drug Targets. 3. pp. 207–44. doi:10.2174/9781681082332116030009. ISBN 978-1-68108-233-2.
- Schreiber KH, Arriola Apelo SI, Yu D, Brinkman JA, Velarde MC, Syed FA, et al. (July 2019). "A novel rapamycin analog is highly selective for mTORC1 in vivo". Nature Communications. 10 (1): 3194. Bibcode:2019NatCo..10.3194S. doi:10.1038/s41467-019-11174-0. PMC 6642166. PMID 31324799.
- Mahoney SJ, Narayan S, Molz L, Berstler LA, Kang SA, Vlasuk GP, Saiah E (February 2018). "A small molecule inhibitor of Rheb selectively targets mTORC1 signaling". Nature Communications. 9 (1): 548. Bibcode:2018NatCo...9..548M. doi:10.1038/s41467-018-03035-z. PMC 5803267. PMID 29416044.
- Johnson SC, Rabinovitch PS, Kaeberlein M (Jan 2013). "mTOR is a key modulator of ageing and age-related disease". Nature. 493 (7432): 338–45. Bibcode:2013Natur.493..338J. doi:10.1038/nature11861. PMC 3687363. PMID 23325216.
External links
- mTORC1+complex,+human at the US National Library of Medicine Medical Subject Headings (MeSH)