IDH2
Isocitrate dehydrogenase [NADP], mitochondrial is an enzyme that in humans is encoded by the IDH2 gene.[5]





Isocitrate dehydrogenases are enzymes that catalyze the oxidative decarboxylation of isocitrate to 2-oxoglutarate. These enzymes belong to two distinct subclasses, one of which utilizes NAD(+) as the electron acceptor and the other NADP(+). Five isocitrate dehydrogenases have been reported: three NAD(+)-dependent isocitrate dehydrogenases, which localize to the mitochondrial matrix, and two NADP(+)-dependent isocitrate dehydrogenases, one of which is mitochondrial and the other predominantly cytosolic. Each NADP(+)-dependent isozyme is a homodimer. The protein encoded by the IDH2 gene is the NADP(+)-dependent isocitrate dehydrogenase found in the mitochondria. It plays a role in intermediary metabolism and energy production. This protein may tightly associate or interact with the pyruvate dehydrogenase complex.[5] Somatic mosaic mutations of this gene have also been found associated to Ollier disease and Maffucci syndrome.[6]
Structure
Isocitrate dehydrogenase is composed of 3 subunits, allosterically regulated, and requires an integrated Mg2+ or Mn2+ ion. The mitochondrial form of IDH, like most isoforms, is a homodimer, in which two identical monomer subunits form one unit. The structure of Mycobacterium tuberculosis IDH-1 bound with NADPH and Mn2+ has been solved by X-ray crystallography. It is a homodimer in which each subunit has a Rossmann fold, and a common top domain of interlocking β sheets. Mtb IDH-1 is most structurally similar to the R132H mutant human IDH found in certain glioblastomas. Similar to human R132H ICDH, Mtb ICDH-1 also catalyzes the formation of α-hydroxyglutarate.[7]
Function
Isocitrate dehydrogenase is a digestive enzyme that is used in the citric acid cycle. Its main function is to catalyze the oxidative decarboxylation of isocitrate into alpha-ketoglutarate. Human isocitrate dehydrogenase regulation is not fully understood however, it is known that NADP and Ca2+ bind in the active site to create three different conformations. These conformations form in the active site and are as follows: a loop is form in the inactive enzyme, a partially unraveled alpha helix in the semi open form, and an alpha helix in the active form.[8]
Clinical significance
The mitochondrial form of IDH2 is correlated with many diseases. Mutations in IDH2 are associated with 2-hydroxyglutaric aciduria, a condition that causes progressive damage to the brain. The major types of this disorder are called D-2-hydroxyglutaric aciduria (D-2-HGA), L-2-hydroxyglutaric aciduria (L-2-HGA), and combined D,L-2-hydroxyglutaric aciduria (D,L-2-HGA). The main features of D-2-HGA are delayed development, seizures, weak muscle tone (hypotonia), and abnormalities in the largest part of the brain (the cerebrum), which controls many important functions such as muscle movement, speech, vision, thinking, emotion, and memory. Researchers have described two subtypes of D-2-HGA, type I and type II. The two subtypes are distinguished by their genetic cause and pattern of inheritance, although they also have some differences in signs and symptoms. Type II tends to begin earlier and often causes more severe health problems than type I. Type II may also be associated with a weakened and enlarged heart (cardiomyopathy), a feature that is typically not found with type I. L-2-HGA particularly affects a region of the brain called the cerebellum, which is involved in coordinating movements. As a result, many affected individuals have problems with balance and muscle coordination (ataxia). Additional features of L-2-HGA can include delayed development, seizures, speech difficulties, and an unusually large head (macrocephaly). Typically, signs and symptoms of this disorder begin during infancy or early childhood. The disorder worsens over time, usually leading to severe disability by early adulthood. Combined D,L-2-HGA causes severe brain abnormalities that become apparent in early infancy. Affected infants have severe seizures, weak muscle tone (hypotonia), and breathing and feeding problems. They usually survive only into infancy or early childhood.[5]
Mutations in the IDH2 gene, along with mutations in the IDH1 gene, are also strongly correlated with the development of glioma, acute myeloid leukemia (AML), chondrosarcoma, intrahepatic cholangiocarcinoma (ICC), and angioimmunoblastic T-cell lymphoma cancers. They also cause D-2-hydroxyglutaric aciduria and Ollier and Maffucci syndromes. IDH2 mutations may allow prolonged survival of glioma and ICC cancer cells, but not AML cells. The reason for this is unknown. Missense mutations in the active site of these IDH2 induce a neo-enzymatic reaction wherein NADPH reduces αKG to D-2-hydroxyglutarate, which accumulates and leads to the inhibition of hypoxia-inducible factor 1α (HIF1α) degradation (inhibition of the HIF prolyl-hydroxylase), as well as changes in epigenetics and extracellular matrix homeostasis. Such mutations also imply less NADPH production capacity.[9] Tumors of various tissue types with IDH1/2 mutations show improved responses to radiation and chemotherapy.[10][11]
Inhibitors of the neomorphic activity of mutant IDH1 and IDH2 are currently in Phase I/II clinical trials for both solid and blood tumors. As IDH1 and IDH2 represent key enzymes within the tricarboxylic acid (TCA) cycle, mutations have significant impact on intermediary metabolism. The loss of some wild-type metabolic activity is an important, potentially deleterious and therapeutically exploitable consequence of oncogenic IDH mutations and requires continued investigation in the future.[12]
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles. [§ 1]
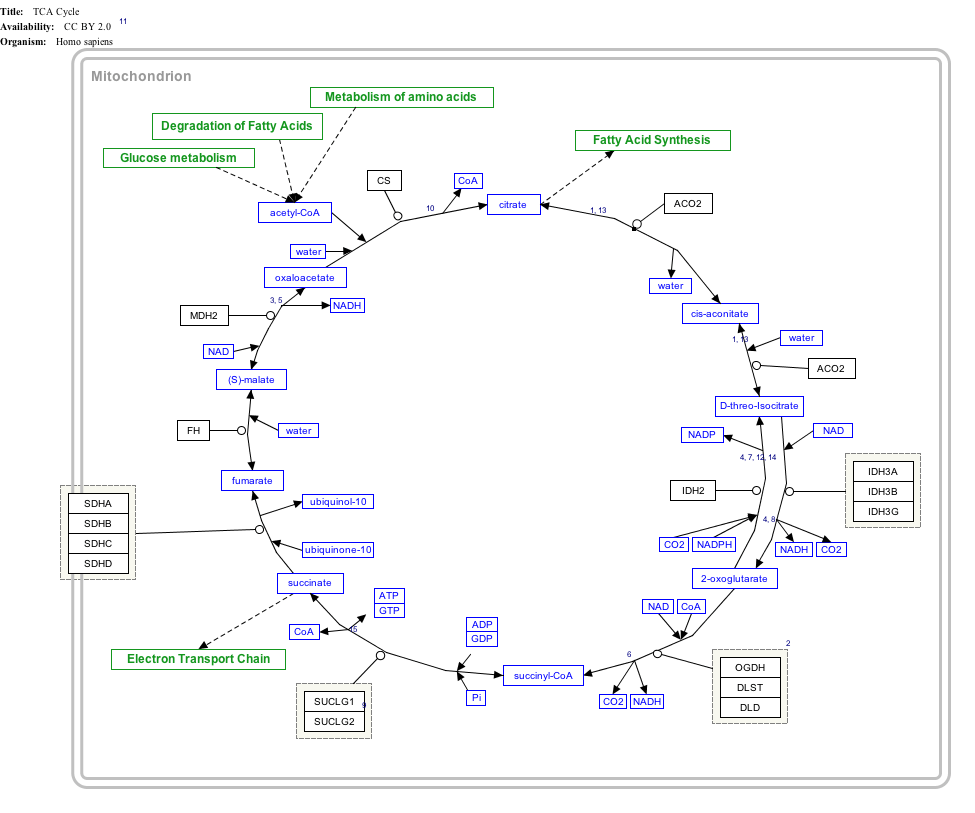
- The interactive pathway map can be edited at WikiPathways: "TCACycle_WP78".
References
- GRCh38: Ensembl release 89: ENSG00000182054 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000030541 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Entrez Gene: IDH2 isocitrate dehydrogenase 2 (NADP+), mitochondrial".
- Amary MF, Damato S, Halai D, Eskandarpour M, Berisha F, Bonar F, et al. (November 2011). "Ollier disease and Maffucci syndrome are caused by somatic mosaic mutations of IDH1 and IDH2". Nature Genetics. 43 (12): 1262–5. doi:10.1038/ng.994. PMID 22057236. S2CID 5592593.
- Quartararo CE, Hazra S, Hadi T, Blanchard JS (March 2013). "Structural, kinetic and chemical mechanism of isocitrate dehydrogenase-1 from Mycobacterium tuberculosis". Biochemistry. 52 (10): 1765–75. doi:10.1021/bi400037w. PMC 3706558. PMID 23409873.
- Xu X, Zhao J, Xu Z, Peng B, Huang Q, Arnold E, Ding J (August 2004). "Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity". The Journal of Biological Chemistry. 279 (32): 33946–57. doi:10.1074/jbc.M404298200. PMID 15173171.
- Molenaar RJ, Radivoyevitch T, Maciejewski JP, van Noorden CJ, Bleeker FE (December 2014). "The driver and passenger effects of isocitrate dehydrogenase 1 and 2 mutations in oncogenesis and survival prolongation". Biochimica et Biophysica Acta (BBA) - Reviews on Cancer. 1846 (2): 326–41. doi:10.1016/j.bbcan.2014.05.004. PMID 24880135.
- Molenaar RJ, Maciejewski JP, Wilmink JW, van Noorden CJ (April 2018). "Wild-type and mutated IDH1/2 enzymes and therapy responses". Oncogene. 37 (15): 1949–1960. doi:10.1038/s41388-017-0077-z. PMC 5895605. PMID 29367755.
- Miyata S, Tominaga K, Sakashita E, Urabe M, Onuki Y, Gomi A, et al. (July 2019). "R132H Clinical Glioma Samples Reveals Suppression of β-oxidation Due to Carnitine Deficiency". Scientific Reports. 9 (1): 9787. doi:10.1038/s41598-019-46217-5. PMC 6611790. PMID 31278288.
- Parker SJ, Metallo CM (August 2015). "Metabolic consequences of oncogenic IDH mutations". Pharmacology & Therapeutics. 152: 54–62. doi:10.1016/j.pharmthera.2015.05.003. PMC 4489982. PMID 25956465.
Further reading
- Bruns GA, Eisenman RE, Gerald PS (1977). "Human mitochondrial NADP-dependent isocitrate dehydrogenase in man-mouse somatic cell hybrids". Cytogenetics and Cell Genetics. 17 (4): 200–11. doi:10.1159/000130713. PMID 11969.
- Shimizu N, Giles RE, Kucherlapati RS, Shimizu Y, Ruddle FH (January 1977). "Somatic cell genetic assignment of the human gene for mitochondrial NADP-linked isocitrate dehydrogenase to the long arm of chromosome 15". Somatic Cell Genetics. 3 (1): 47–60. doi:10.1007/BF01550986. PMID 564083. S2CID 32512064.
- Champion MJ, Brown JA, Shows TB (1979). "Assignment of cytoplasmic alpha-mannosidase (MANA) and confirmation of mitochondrial isocitrate dehydrogenase (IDHM) to the q11 leads to qter region of chromosome 15 in man". Cytogenetics and Cell Genetics. 22 (1–6): 498–502. doi:10.1159/000131007. PMID 752528.
- Grzeschik KH (September 1976). "Assignment of a gene for human mitochondrial isocitrate dehydrogenase (ICD-M, EC 1.1.1.41) to chromosome 15". Human Genetics. 34 (1): 23–8. doi:10.1007/BF00284430. PMID 965003. S2CID 5544877.
- Klimek J, Boguslawski W, Tialowska B, Zelewski L (1976). "Regulation of progesterone biosynthesis in human placental mitochondria by Krebs cycle metabolites". Acta Biochimica Polonica. 23 (2–3): 185–92. PMID 970033.
- Chamberlain KG, Penington DG (February 1988). "Monoamine oxidase and other mitochondrial enzymes in density subpopulations of human platelets". Thrombosis and Haemostasis. 59 (1): 29–33. doi:10.1055/s-0038-1642560. PMID 3363531.
- Maruyama K, Sugano S (January 1994). "Oligo-capping: a simple method to replace the cap structure of eukaryotic mRNAs with oligoribonucleotides". Gene. 138 (1–2): 171–4. doi:10.1016/0378-1119(94)90802-8. PMID 8125298.
- Luo H, Shan X, Wu J (March 1996). "Expression of human mitochondrial NADP-dependent isocitrate dehydrogenase during lymphocyte activation". Journal of Cellular Biochemistry. 60 (4): 495–507. doi:10.1002/(SICI)1097-4644(19960315)60:4<495::AID-JCB6>3.0.CO;2-N. PMID 8707889.
- Oh IU, Inazawa J, Kim YO, Song BJ, Huh TL (November 1996). "Assignment of the human mitochondrial NADP(+)-specific isocitrate dehydrogenase (IDH2) gene to 15q26.1 by in situ hybridization". Genomics. 38 (1): 104–6. doi:10.1006/geno.1996.0602. PMID 8954790.
- Suzuki Y, Yoshitomo-Nakagawa K, Maruyama K, Suyama A, Sugano S (October 1997). "Construction and characterization of a full length-enriched and a 5'-end-enriched cDNA library". Gene. 200 (1–2): 149–56. doi:10.1016/S0378-1119(97)00411-3. PMID 9373149.
- Gevaert K, Goethals M, Martens L, Van Damme J, Staes A, Thomas GR, Vandekerckhove J (May 2003). "Exploring proteomes and analyzing protein processing by mass spectrometric identification of sorted N-terminal peptides". Nature Biotechnology. 21 (5): 566–9. doi:10.1038/nbt810. PMID 12665801. S2CID 23783563.
- Foster LJ, Rudich A, Talior I, Patel N, Huang X, Furtado LM, et al. (January 2006). "Insulin-dependent interactions of proteins with GLUT4 revealed through stable isotope labeling by amino acids in cell culture (SILAC)". Journal of Proteome Research. 5 (1): 64–75. doi:10.1021/pr0502626. PMID 16396496.
- Kil IS, Kim SY, Lee SJ, Park JW (October 2007). "Small interfering RNA-mediated silencing of mitochondrial NADP+-dependent isocitrate dehydrogenase enhances the sensitivity of HeLa cells toward tumor necrosis factor-alpha and anticancer drugs". Free Radical Biology & Medicine. 43 (8): 1197–207. doi:10.1016/j.freeradbiomed.2007.07.009. PMID 17854715.
External links
- Overview of all the structural information available in the PDB for UniProt: P48735 (Human Isocitrate dehydrogenase [NADP], mitochondrial) at the PDBe-KB.
- Overview of all the structural information available in the PDB for UniProt: P54071 (Mouse Isocitrate dehydrogenase [NADP], mitochondrial) at the PDBe-KB.
PDB gallery | |
|---|---|
|
