CCR5
C-C chemokine receptor type 5, also known as CCR5 or CD195, is a protein on the surface of white blood cells that is involved in the immune system as it acts as a receptor for chemokines.[5]
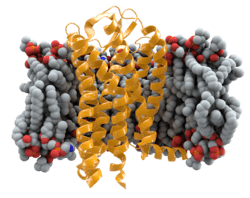
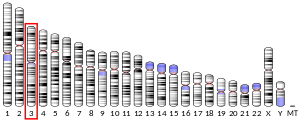

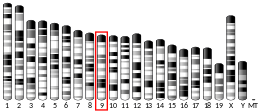

In humans, the CCR5 gene that encodes the CCR5 protein is located on the short (p) arm at position 21 on chromosome 3. Certain populations have inherited the Delta 32 mutation, resulting in the genetic deletion of a portion of the CCR5 gene. Homozygous carriers of this mutation are resistant to M-tropic strains of HIV-1 infection.[6][7][8][9][10][11]
Function
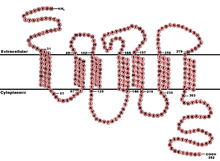
The CCR5 protein belongs to the beta chemokine receptors family of integral membrane proteins.[12][13] It is a G protein–coupled receptor[12] which functions as a chemokine receptor in the CC chemokine group.
CCR5's cognate ligands include CCL3, CCL4 (also known as MIP 1α and 1β, respectively), and CCL3L1.[14][15] CCR5 furthermore interacts with CCL5 (a chemotactic cytokine protein also known as RANTES).[14][16][17]
CCR5 is predominantly expressed on T cells, macrophages, dendritic cells, eosinophils, microglia and a subpopulation of either breast or prostate cancer cells.[18][19] The expression of CCR5 is selectively induced during the cancer transformation process and is not expressed in normal breast or prostate epithelial cells. Approximately 50% of human breast cancer expressed CCR5, primarily in triple negative breast cancer.[18] CCR5 inhibitors blocked the migration and metastasis of breast and prostate cancer cells that expressed CCR5, suggesting that CCR5 may function as a new therapeutic target.[18][19][20] Recent studies suggest that CCR5 is expressed in a subset of cancer cells with characteristics of cancer stem cells, which are known to drive therapy resistance, and that CCR5 inhibitors enhanced the number of cells killed by current chemotherapy.[21] It is likely that CCR5 plays a role in inflammatory responses to infection, though its exact role in normal immune function is unclear. Regions of this protein are also crucial for chemokine ligand binding, the functional response of the receptor, and HIV co-receptor activity.[22]
HIV
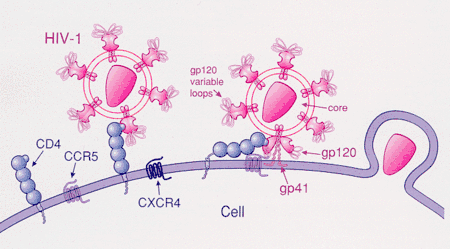
HIV-1 most commonly uses the chemokine receptors CCR5 and/or CXCR4 as co-receptors to enter target immunological cells.[23] These receptors are located on the surface of host immune cells whereby they provide a method of entry for the HIV-1 virus to infect the cell.[24] The HIV-1 envelope glycoprotein structure is essential in enabling the viral entry of HIV-1 into a target host cell.[24] The envelope glycoprotein structure consists of two protein subunits cleaved from a Gp160 protein precursor encoded for by the HIV-1 env gene: the Gp120 external subunit and the Gp41 transmembrane subunit.[24] This envelope glycoprotein structure is arranged into a spike-like structure located on the surface of the virion and consists of a trimer of Gp120-Gp41 hetero-dimers.[24] The Gp120 envelope protein is a chemokine mimic.[23] Though it lacks the unique structure of a chemokine, it is still capable of binding to the CCR5 and CXCR4 chemokine receptors.[23] During HIV-1 infection, the Gp120 envelope glycoprotein subunit binds to a CD4 glycoprotein and a HIV-1 co-receptor expressed on a target cell, forming a heterotrimeric complex.[23] The formation of this complex stimulates the release of a fusogenic peptide, causing the viral membrane to fuse with the membrane of the target host cell.[23] Because binding to CD4 alone can sometimes result in gp120 shedding, gp120 must next bind to co-receptor CCR5 in order for fusion to proceed. The tyrosine-sulfated amino terminus of this co-receptor is the "essential determinant" of binding to the gp120 glycoprotein.[25] The co-receptor also recognizes the V1-V2 region of gp120 and the bridging sheet (an antiparallel, 4-stranded β sheet that connects the inner and outer domains of gp120). The V1-V2 stem can influence "co-receptor usage through its peptide composition as well as by the degree of N-linked glycosylation." Unlike V1-V2 however, the V3 loop is highly variable and thus is the most important determinant of co-receptor specificity.[25] The normal ligands for this receptor, RANTES, MIP-1β, and MIP-1α, are able to suppress HIV-1 infection in vitro. In individuals infected with HIV, CCR5-using viruses are the predominant species isolated during the early stages of viral infection,[26] suggesting that these viruses may have a selective advantage during transmission or the acute phase of disease. Moreover, at least half of all infected individuals harbor only CCR5-using viruses throughout the course of infection.
CCR5 is the primary co-receptor used by gp120 sequentially with CD4. This bind results in gp41, the other protein product of gp160, to be released from its metastable conformation and insert itself into the membrane of the host cell. Although it has not been confirmed, binding of gp120-CCR5 involves two crucial steps: 1) The tyrosine-sulfated amino terminus of this co-receptor is an "essential determinant" of binding to gp120 (as stated previously) 2) Following step 1., there must be reciprocal action (synergy, intercommunication) between gp120 and the CCR5 transmembrane domains.[25]
CCR5 is essential for the spread of the R5-strain of the HIV-1 virus.[27] Knowledge of the mechanism by which this strain of HIV-1 mediates infection has prompted research into the development of therapeutic interventions to block CCR5 function.[28] A number of new experimental HIV drugs, called CCR5 receptor antagonists, have been designed to interfere with binding between the Gp120 envelope protein and the HIV co-receptor CCR5.[27] These experimental drugs include PRO140 (CytoDyn), Vicriviroc (Phase III trials were cancelled in July 2010) (Schering Plough), Aplaviroc (GW-873140) (GlaxoSmithKline) and Maraviroc (UK-427857) (Pfizer). Maraviroc was approved for use by the FDA in August 2007.[27] It is the only one thus far approved by the FDA for clinical use, thus becoming the first CCR5 inhibitor.[25] A problem of this approach is that, while CCR5 is the major co-receptor by which HIV infects cells, it is not the only such co-receptor. It is possible that under selective pressure HIV will evolve to use another co-receptor. However, examination of viral resistance to AD101, molecular antagonist of CCR5, indicated that resistant viruses did not switch to another co-receptor (CXCR4), but persisted in using CCR5: they either bound to alternative domains of CCR5 or to the receptor at a higher affinity. However, because there is still another co-receptor available, it is probable that lacking the CCR5 gene does not make one immune to the virus; it would simply be more challenging for the individual to contract it. Also, the virus still has access to CD4. Unlike CCR5, which is not required (as evidenced by those living healthy lives even when lacking the gene as a result of the delta32 mutation), CD4 is critical in the body's immune defense system.[29] Even without the availability of either co-receptor (even CCR5), the virus can still invade cells if gp41 were to go through an alteration (including its cytoplasmic tail) that resulted in the independence of CD4 without the need of CCR5 and/or CXCR4 as a doorway.[30]
Cancer
Expression of CCR5 is induced in breast and prostate epithelial cells upon transformation. The induction of CCR5 expression promotes cellular invasion, migration, and metastasis. The induction of metastasis involves homing to the metastatic site. CCR5 inhibitors including maraviroc and leronlimab have been shown to block lung metastasis of human breast cancer cell lines.[18] In preclinical studies of immune competent mice CCR5 inhibitors blocked metastasis to the bones and brain.[19] CCR5 inhibitors also reduce the infiltration of tumor associated macrophages.[31] A Phase 1 clinical study of a CCR5 inhibitor in heavily-pretreated patients with metastatic colon cancer demonstrated an objective clinical response and reduction in metastatic tumor burden.[32]
Brain
Increased levels of CCR5 are part of the inflammatory response to stroke. Blocking CCR5 with Maraviroc (a drug approved for HIV) may enhance recovery after stroke.[33][34]
In the developing brain, chemokines such as CCR5 influence neuronal migration and connection. After stroke, they seem to decrease the number of connection sites on neurons near the damage.[33]
CCR5-Δ32
CCR5-Δ32 (or CCR5-D32 or CCR5 delta 32) is an allele of CCR5.[35][36]
CCR5 Δ32 is a 32-base-pair deletion that introduces a premature stop codon into the CCR5 receptor locus, resulting in a nonfunctional receptor.[37][38] CCR5 is required for M-tropic HIV-1 virus entry.[39] Individuals homozygous (denoted Δ32/Δ32) for CCR5 Δ32 do not express functional CCR5 receptors on their cell surfaces and are resistant to HIV-1 infection, despite multiple high-risk exposures.[39] Individuals heterozygous (+/Δ32) for the mutant allele have a greater than 50% reduction in functional CCR5 receptors on their cell surfaces due to dimerization between mutant and wild-type receptors that interferes with transport of CCR5 to the cell surface.[40] Heterozygote carriers are resistant to HIV-1 infection relative to wild types and when infected, heterozygotes exhibit reduced viral loads and a 2-3-year-slower progression to AIDS relative to wild types.[37][39][41] Heterozygosity for this mutant allele also has shown to improve one's virological response to anti-retroviral treatment.[42] CCR5 Δ32 has an (heterozygote) allele frequency of 10% in Europe, and a homozygote frequency of 1%.
Recent research indicates that CCR5 Δ32 enhances cognition and memory. In 2016, researchers showed that removing the CCR5 gene from mice significantly improved their memory.[43] CCR5 is a powerful suppressor for neuronal plasticity, learning, and memory; CCR5 over-activation by viral proteins may contribute to HIV-associated cognitive deficits.[44]
Evolutionary history and age of the allele
The CCR5 Δ32 allele is notable for its recent origin, unexpectedly high frequency, and distinct geographic distribution,[45] which together suggest that (a) it arose from a single mutation, and (b) it was historically subject to positive selection.
Two studies have used linkage analysis to estimate the age of the CCR5 Δ32 deletion, assuming that the amount of recombination and mutation observed on genomic regions surrounding the CCR5 Δ32 deletion would be proportional to the age of the deletion.[36][46] Using a sample of 4000 individuals from 38 ethnic populations, Stephens et al. estimated that the CCR5-Δ32 deletion occurred 700 years ago (275-1875, 95% confidence interval). Another group, Libert et al. (1998), used microsatellite mutations to estimate the age of the CCR5 Δ32 mutation to be 2100 years (700-4800, 95% confidence interval). On the basis of observed recombination events, they estimated the age of the mutation to be 2250 years (900-4700, 95% confidence interval).[46] A third hypothesis relies on the north-to-south gradient of allele frequency in Europe, which shows that the highest allele frequency occurred in the Nordic countries and lowest allele frequency in southern Europe. Because the Vikings historically occupied these countries, it may be possible that the allele spread throughout Europe due to the Viking dispersal in the 8th to 10th centuries.[47] Vikings were later replaced by the Varangians in Russia, which may have contributed to the observed east-to-west cline of allele frequency.[45][47]
HIV-1 was initially transmitted from chimpanzees (Pan troglodytes) to humans in the early 1900s in Southeast Cameroon, Africa,[48] through exposure to infected blood and body fluids while butchering bushmeat.[49] However, HIV-1 was effectively absent from Europe until the 1980s.[50] Therefore, given the average age of roughly 1000 years for the CCR5-Δ32 allele, it can be established that HIV-1 did not exert selection pressure on the human population for long enough to achieve the current frequencies.[45] Hence, other pathogens have been suggested as agents of positive selection for CCR5 Δ32, including bubonic plague (Yersinia pestis) and smallpox (Variola major). Other data suggest that the allele frequency experienced negative selection pressure as a result of pathogens that became more widespread during Roman expansion.[51] The idea that negative selection played a role in the allele's low frequency is also supported by experiments using knockout mice and Influenza A, which demonstrated that the presence of the CCR5 receptor is important for efficient response to a pathogen.[52][53]
Evidence for a single mutation
Several lines of evidence suggest that the CCR5 Δ32 allele evolved only once.[45] First, CCR5 Δ32 has a relatively high frequency in several different European populations but is comparatively absent in Asian, Middle Eastern and American Indian populations,[36] suggesting that a single mutation occurred after divergence of Europeans from their African ancestor.[36][37][54] Second, genetic linkage analysis indicates that the mutation occurs on a homogenous genetic background, implying that inheritance of the mutation occurred from a common ancestor.[46] This was demonstrated by showing that the CCR5 Δ32 allele is in strong linkage disequilibrium with highly polymorphic microsatellites. More than 95% of CCR5 Δ32 chromosomes also carried the IRI3.1-0 allele, while 88% carried the IRI3.2 allele. By contrast, the microsatellite markers IRI3.1-0 and IRI3.2-0 were found in only 2 or 1.5% of chromosomes carrying a wild-type CCR5 allele.[46] This evidence of linkage disequilibrium supports the hypothesis that most, if not all, CCR5 Δ32 alleles arose from a single mutational event. Finally, the CCR5 Δ32 allele has a unique geographical distribution indicating a single Northern origin followed by migration. A study measuring allele frequencies in 18 European populations found a North-to-South gradient, with the highest allele frequencies in Finnish and Mordvinian populations (16%), and the lowest in Sardinia (4%).[46]
Positive selection
In the absence of selection, a single mutation would take an estimated 127,500 years to rise to a population frequency of 10%.[36] Estimates based on genetic recombination and mutation rates place the age of the allele between 1000 and 2000 years. This discrepancy is a signature of positive selection.
It is estimated that HIV-1 entered the human population in Africa in the early 1900s;[48] however symptomatic infections were not reported until the 1980s. The HIV-1 epidemic is therefore far too young to be the source of positive selection that drove the frequency of CCR5 Δ32 from zero to 10% in 2000 years. Stephens, et al. (1998), suggest that bubonic plague (Yersinia pestis) had exerted positive selective pressure on CCR5 Δ32.[36] This hypothesis was based on the timing and severity of the Black Death pandemic, which killed 30% of the European population of all ages between 1346 and 1352.[55] After the Black Death, there were less severe, intermittent epidemics. Individual cities experienced high mortality, but overall mortality in Europe was only a few percent.[55][56][57] In 1655-1656 a second pandemic called the "Great Plague" killed 15-20% of Europe's population.[55][58] Importantly, the plague epidemics were intermittent. Bubonic plague is a zoonotic disease, primarily infecting rodents, spread by fleas, and only occasionally infecting humans.[59] Human-to-human infection of bubonic plague does not occur, though it can occur in pneumonic plague, which infects the lungs.[60] Only when the density of rodents is low are infected fleas forced to feed on alternative hosts such as humans, and under these circumstances a human epidemic may occur.[59] Based on population genetic models, Galvani and Slatkin (2003) argue that the intermittent nature of plague epidemics did not generate a sufficiently strong selective force to drive the allele frequency of CCR5 Δ32 to 10% in Europe.[35]
To test this hypothesis, Galvani and Slatkin (2003) modeled the historical selection pressures produced by plague and smallpox.[35] Plague was modeled according to historical accounts,[61][62] while age-specific smallpox mortality was gleaned from the age distribution of smallpox burials in York (England) between 1770 and 1812.[56] Smallpox preferentially infects young, pre-reproductive members of the population since they are the only individuals who are not immunized or dead from past infection. Because smallpox preferentially kills pre-reproductive members of a population, it generates stronger selective pressure than plague.[35] Unlike plague, smallpox does not have an animal reservoir and is only transmitted from human to human.[63][64] The authors calculated that if plague were selecting for CCR5 Δ32, the frequency of the allele would still be less than 1%, while smallpox has exerted a selective force sufficient to reach 10%.
The hypothesis that smallpox exerted positive selection for CCR5 Δ32 is also biologically plausible, since poxviruses, like HIV, enter white blood cells using chemokine receptors.[65] By contrast, Yersinia pestis is a bacterium with a very different biology.
Although Europeans are the only group to have subpopulations with a high frequency of CCR5 Δ32, they are not the only population that has been subject to selection by smallpox, which had a worldwide distribution before it was declared eradicated in 1980. The earliest unmistakable descriptions of smallpox appear in the 5th century A.D. in China, the 7th century A.D. in India and the Mediterranean, and the 10th century A.D. in southwestern Asia.[64] By contrast, the CCR5 Δ32 mutation is found only in European, West Asian, and North African populations.[66] The anomalously high frequency of CCR5 Δ32 in these populations appears to require both a unique origin in Northern Europe and subsequent selection by smallpox.
Potential costs
CCR5 Δ32 can be beneficial to the host in some infections (e.g., HIV-1, possibly smallpox), but detrimental in others (e.g., tick-borne encephalitis, West Nile virus). Whether CCR5 function is helpful or harmful in the context of a given infection depends on a complex interplay between the immune system and the pathogen.
In general, research suggests that the CCR5 Δ32 mutation may play a deleterious role in post-infection inflammatory processes, which can injure tissue and create further pathology.[67] The best evidence for this proposed antagonistic pleiotropy is found in flavivirus infections. In general many viral infections are asymptomatic or produce only mild symptoms in the vast majority of the population. However, certain unlucky individuals experience a particularly destructive clinical course, which is otherwise unexplained but appears to be genetically mediated. Patients homozygous for CCR5 Δ32 were found to be at higher risk for a neuroinvasive form of tick-borne encephalitis (a flavivirus).[68] In addition, functional CCR5 may be required to prevent symptomatic disease after infection with West Nile virus, another flavivirus; CCR5 Δ32 was associated with early symptom development and more pronounced clinical manifestations after infection with West Nile virus.[69]
This finding in humans confirmed a previously observed experiment in an animal model of CCR5 Δ32 homozygosity. After infection with West Nile Virus, CCR5 Δ32 mice had markedly increased viral titers in the central nervous system and had increased mortality[70] compared with that of wild-type mice, thus suggesting that CCR5 expression was necessary to mount a strong host defense against West Nile virus.
Medical applications
A genetic approach involving intrabodies that block CCR5 expression has been proposed as a treatment for HIV-1 infected individuals.[71] When T-cells modified so they no longer express CCR5 were mixed with unmodified T-cells expressing CCR5 and then challenged by infection with HIV-1, the modified T-cells that do not express CCR5 eventually take over the culture, as HIV-1 kills the non-modified T-cells. This same method might be used in vivo to establish a virus-resistant cell pool in infected individuals.[71]
This hypothesis was tested in an AIDS patient who had also developed myeloid leukemia, and was treated with chemotherapy to suppress the cancer. A bone marrow transplant containing stem cells from a matched donor was then used to restore the immune system. However, the transplant was performed from a donor with 2 copies of CCR5-Δ32 mutation gene. After 600 days, the patient was healthy and had undetectable levels of HIV in the blood and in examined brain and rectal tissues.[7][72] Before the transplant, low levels of HIV X4, which does not use the CCR5 receptor, were also detected. Following the transplant, however, this type of HIV was not detected either.[7] However, this outcome is consistent with the observation that cells expressing the CCR5-Δ32 variant protein lack both the CCR5 and CXCR4 receptors on their surfaces, thereby conferring resistance to a broad range of HIV variants including HIVX4.[73] After over six years, the patient has maintained the resistance to HIV and has been pronounced cured of the HIV infection.[8]
Enrollment of HIV-positive patients in a clinical trial was started in 2009 in which the patients' cells were genetically modified with a zinc finger nuclease to carry the CCR5-Δ32 trait and then reintroduced into the body as a potential HIV treatment.[74][75] Results reported in 2014 were promising.[11]
Inspired by the first person ever to be cured of HIV, The Berlin Patient, StemCyte began collaborations with Cord blood banks worldwide to systematically screen Umbilical cord blood samples for the CCR5 mutation beginning in 2011.[76][77][78]
In November 2018, Jiankui He announced that he had edited two human embryos, to attempt to disable the gene for CCR5, which codes for a receptor that HIV uses to enter cells. He said that twin girls, Lulu and Nana, had been born a few weeks earlier, and that the girls still carried functional copies of CCR5 along with disabled CCR5 (mosaicism), hence being still vulnerable to HIV. The work was widely condemned as unethical, dangerous, and premature.[79][80]
See also
- Discovery and development of CCR5 receptor antagonists
- Entry inhibitor
- HIV tropism
- Stephen Crohn
- HIV immunity
References
- GRCh38: Ensembl release 89: ENSG00000160791 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000079227 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Jiao X, Nawab O, Patel T, Kossenkov AV, Halama N, Jaeger D, Pestell RG (Oct 2019). "Recent Advances Targeting CCR5 for Cancer and Its Role in Immuno-Oncology". Cancer Research. 79 (19): 4801–4807. doi:10.1158/0008-5472.CAN-19-1167. PMC 6810651. PMID 31292161.
- de Silva E, Stumpf MP (Dec 2004). "HIV and the CCR5-Delta32 resistance allele". FEMS Microbiology Letters. 241 (1): 1–12. doi:10.1016/j.femsle.2004.09.040. PMID 15556703.
- Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K, Schneider T, Hofmann J, Kücherer C, Blau O, Blau IW, Hofmann WK, Thiel E (Feb 2009). "Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation". The New England Journal of Medicine. 360 (7): 692–8. doi:10.1056/NEJMoa0802905. PMID 19213682.
- Allers K, Hütter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, Schneider T (Mar 2011). "Evidence for the cure of HIV infection by CCR5Δ32/Δ32 stem cell transplantation". Blood. 117 (10): 2791–9. doi:10.1182/blood-2010-09-309591. PMID 21148083.
- Zhen A, Kitchen S (Jan 2014). "Stem-cell-based gene therapy for HIV infection". Viruses. 6 (1): 1–12. doi:10.3390/v6010001. PMC 3917429. PMID 24368413.
- Kay MA, Walker BD (Mar 2014). "Engineering cellular resistance to HIV". The New England Journal of Medicine. 370 (10): 968–9. doi:10.1056/NEJMe1400593. PMID 24597871.
- Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, et al. (Mar 2014). "Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV". The New England Journal of Medicine. 370 (10): 901–10. doi:10.1056/NEJMoa1300662. PMC 4084652. PMID 24597865.
- "CCR5 - chemokine (C-C motif) receptor 5 (gene/pseudogene)". Genetics Home Reference.
- Samson M, Labbe O, Mollereau C, Vassart G, Parmentier M (Mar 1996). "Molecular cloning and functional expression of a new human CC-chemokine receptor gene". Biochemistry. 35 (11): 3362–7. doi:10.1021/bi952950g. PMID 8639485.
- Struyf S, Menten P, Lenaerts JP, Put W, D'Haese A, De Clercq E, Schols D, Proost P, Van Damme J (Jul 2001). "Diverging binding capacities of natural LD78beta isoforms of macrophage inflammatory protein-1alpha to the CC chemokine receptors 1, 3 and 5 affect their anti-HIV-1 activity and chemotactic potencies for neutrophils and eosinophils". European Journal of Immunology. 31 (7): 2170–8. doi:10.1002/1521-4141(200107)31:7<2170::AID-IMMU2170>3.0.CO;2-D. PMID 11449371.
- Miyakawa T, Obaru K, Maeda K, Harada S, Mitsuya H (Feb 2002). "Identification of amino acid residues critical for LD78beta, a variant of human macrophage inflammatory protein-1alpha, binding to CCR5 and inhibition of R5 human immunodeficiency virus type 1 replication". The Journal of Biological Chemistry. 277 (7): 4649–55. doi:10.1074/jbc.M109198200. PMID 11734558.
- Slimani H, Charnaux N, Mbemba E, Saffar L, Vassy R, Vita C, Gattegno L (Oct 2003). "Interaction of RANTES with syndecan-1 and syndecan-4 expressed by human primary macrophages". Biochimica et Biophysica Acta (BBA) - Biomembranes. 1617 (1–2): 80–8. doi:10.1016/j.bbamem.2003.09.006. PMID 14637022.
- Proudfoot AE, Fritchley S, Borlat F, Shaw JP, Vilbois F, Zwahlen C, Trkola A, Marchant D, Clapham PR, Wells TN (Apr 2001). "The BBXB motif of RANTES is the principal site for heparin binding and controls receptor selectivity". The Journal of Biological Chemistry. 276 (14): 10620–6. doi:10.1074/jbc.M010867200. PMID 11116158.
- Velasco-Velázquez M, Jiao X, De La Fuente M, Pestell TG, Ertel A, Lisanti MP, Pestell RG (Aug 2012). "CCR5 antagonist blocks metastasis of basal breast cancer cells". Cancer Research. 72 (15): 3839–50. doi:10.1158/0008-5472.CAN-11-3917. PMID 22637726.
- Sicoli D, Jiao X, Ju X, Velasco-Velazquez M, Ertel A, Addya S, Li Z, Andò S, Fatatis A, Paudyal B, Cristofanilli M, Thakur ML, Lisanti MP, Pestell RG (Dec 2014). "CCR5 receptor antagonists block metastasis to bone of v-Src oncogene-transformed metastatic prostate cancer cell lines". Cancer Research. 74 (23): 7103–14. doi:10.1158/0008-5472.CAN-14-0612. PMC 4294544. PMID 25452256.
- Velasco-Velázquez M, Xolalpa W, Pestell RG (Nov 2014). "CCL5/CCR5 in breast cancer". Expert Opin Ther Targets. 18 (11): 1265–75. doi:10.1517/14728222.2014.949238. PMID 25256399.
- Jiao X, Velasco-Velázquez MA, Wang M, Li Z, Rui H, Peck AR, Korkola JE, Chen X, Xu S, DuHadaway JB, Guerrero-Rodriguez S, Addya S, Sicoli D, Mu Z, Zhang G, Stucky A, Zhang X, Cristofanilli M, Fatatis A, Gray JW, Zhong JF, Prendergast GC, Pestell RG (Apr 2018). "CCR5 Governs DNA Damage Repair and Breast Cancer Stem Cell Expansion". Cancer Res. 78 (7): 1657–71. doi:10.1158/0008-5472.CAN-17-0915. PMC 6331183. PMID 29358169.
- Barmania F, Pepper MS (2013). "C-C chemokine receptor type five (CCR5): An emerging target for the control of HIV infection". Applied & Translational Genomics. 2 (a): 3–16. doi:10.1016/j.atg.2013.05.004. PMC 5133339. PMID 27942440.
- Murphy PM (Feb 2001). "Viral exploitation and subversion of the immune system through chemokine mimicry". Nature Immunology. 2 (2): 116–22. doi:10.1038/84214. PMID 11175803.
- Alkhatib G (Mar 2009). "The biology of CCR5 and CXCR4". Current Opinion in HIV and AIDS. 4 (2): 96–103. doi:10.1097/COH.0b013e328324bbec. PMC 2718543. PMID 19339947.
- "CCR5 Inhibitors and HIV".
- Anderson J, Akkina R (Sep 2007). "Complete knockdown of CCR5 by lentiviral vector-expressed siRNAs and protection of transgenic macrophages against HIV-1 infection". Gene Therapy. 14 (17): 1287–97. doi:10.1038/sj.gt.3302958. PMID 17597795.
- Lieberman-Blum SS, Fung HB, Bandres JC (Jul 2008). "Maraviroc: a CCR5-receptor antagonist for the treatment of HIV-1 infection". Clinical Therapeutics. 30 (7): 1228–50. doi:10.1016/s0149-2918(08)80048-3. PMID 18691983.
- Nazari R, Joshi S (Aug 2008). "CCR5 as target for HIV-1 gene therapy". Current Gene Therapy. 8 (4): 264–72. doi:10.2174/156652308785160674. PMID 18691022.
- "Understanding Genetics". The Tech Museum of Innovation, San Jose, CA.
- Taylor BM, Foulke JS, Flinko R, Heredia A, DeVico A, Reitz M (Jun 2008). "An alteration of human immunodeficiency virus gp41 leads to reduced CCR5 dependence and CD4 independence". Journal of Virology. 82 (11): 5460–71. doi:10.1128/JVI.01049-07. PMC 2395218. PMID 18353949.
- Frankenberger C, Rabe D, Bainer R, Sankarasharma D, Chada K, Krausz T, Gilad Y, Becker L, Rosner MR (Oct 2015). "Metastasis Suppressors Regulate the Tumor Microenvironment by Blocking Recruitment of Prometastatic Tumor-Associated Macrophages". Cancer Res. 75 (19): 4063–73. doi:10.1158/0008-5472.CAN-14-3394. PMC 4592465. PMID 26238785.
- Halama N, Zoernig I, Berthel A, Kahlert C, Klupp F, Suarez-Carmona M, Suetterlin T, Brand K, Krauss J, Lasitschka F, Lerchl T, Luckner-Minden C, Ulrich A, Koch M, Weitz J, Schneider M, Buechler MW, Zitvogel L, Herrmann T, Benner A, Kunz C, Luecke S, Springfeld C, Grabe N, Falk CS, Jaeger D (Apr 2016). "Tumoral Immune Cell Exploitation in Colorectal Cancer Metastases Can Be Targeted Effectively by Anti-CCR5 Therapy in Cancer Patients". Cancer Cell. 29 (4): 587–601. doi:10.1016/j.ccell.2016.03.005. PMID 27070705.
- "HIV drug could improve recovery after stroke". ScienceMag. 2019-02-21. Retrieved 2019-02-22.
- Joy MT, Assayag EB, Shabashov-Stone D, Liraz-Zaltsman S, Mazzitelli J, Arenas M, Abduljawad N, Kliper E, Korczyn AD, Thareja NS, Kesner EL, Zhou M, Huang S, Silva TK, Katz N, Bornstein NM, Silva AJ, Shohami E, Carmichael ST (2019-02-21). "CCR5 Is a Therapeutic Target for Recovery after Stroke and Traumatic Brain Injury". Cell. 176 (5): 1143–1157. doi:10.1016/j.cell.2019.01.044. PMID 30794775.
- Galvani AP, Slatkin M (Dec 2003). "Evaluating plague and smallpox as historical selective pressures for the CCR5-Delta 32 HIV-resistance allele". Proceedings of the National Academy of Sciences of the United States of America. 100 (25): 15276–9. Bibcode:2003PNAS..10015276G. doi:10.1073/pnas.2435085100. PMC 299980. PMID 14645720.
- Stephens JC, Reich DE, Goldstein DB, Shin HD, Smith MW, Carrington M, et al. (Jun 1998). "Dating the origin of the CCR5-Delta32 AIDS-resistance allele by the coalescence of haplotypes". American Journal of Human Genetics. 62 (6): 1507–15. doi:10.1086/301867. PMC 1377146. PMID 9585595.
- Dean M, Carrington M, Winkler C, Huttley GA, Smith MW, Allikmets R, et al. (Sep 1996). "Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study". Science. 273 (5283): 1856–62. Bibcode:1996Sci...273.1856D. doi:10.1126/science.273.5283.1856. PMID 8791590.
- Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M (Aug 1996). "Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene". Nature. 382 (6593): 722–5. Bibcode:1996Natur.382..722S. doi:10.1038/382722a0. PMID 8751444.
- Liu R, Paxton WA, Choe S, Ceradini D, Martin SR, Horuk R, MacDonald ME, Stuhlmann H, Koup RA, Landau NR (Aug 1996). "Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection". Cell. 86 (3): 367–77. doi:10.1016/s0092-8674(00)80110-5. PMID 8756719.
- Benkirane M, Jin DY, Chun RF, Koup RA, Jeang KT (Dec 1997). "Mechanism of transdominant inhibition of CCR5-mediated HIV-1 infection by ccr5delta32". The Journal of Biological Chemistry. 272 (49): 30603–6. doi:10.1074/jbc.272.49.30603. PMID 9388191.
- Michael NL, Louie LG, Rohrbaugh AL, Schultz KA, Dayhoff DE, Wang CE, Sheppard HW (Oct 1997). "The role of CCR5 and CCR2 polymorphisms in HIV-1 transmission and disease progression". Nature Medicine. 3 (10): 1160–2. doi:10.1038/nm1097-1160. PMID 9334732.
- Laurichesse JJ, Persoz A, Theodorou I, Rouzioux C, Delfraissy JF, Meyer L (May 2007). "Improved virological response to highly active antiretroviral therapy in HIV-1-infected patients carrying the CCR5 Delta32 deletion". HIV Medicine. 8 (4): 213–9. doi:10.1111/j.1468-1293.2007.00455.x. PMID 17461848.
- "China's CRISPR twins might have had their brains inadvertently enhanced". Technology Review. Retrieved 2019-02-22.
- Zhou M, Greenhill S, Huang S, Silva TK, Sano Y, Wu S, Cai Y, Nagaoka Y, Sehgal M, Cai DJ, Lee YS, Fox K, Silva AJ (2016-12-20). "CCR5 is a suppressor for cortical plasticity and hippocampal learning and memory". eLife. 5. doi:10.7554/eLife.20985. PMC 5213777. PMID 27996938.
- Galvani AP, Novembre J (Feb 2005). "The evolutionary history of the CCR5-Delta32 HIV-resistance mutation". Microbes and Infection / Institut Pasteur. 7 (2): 302–9. doi:10.1016/j.micinf.2004.12.006. PMID 15715976.
- Libert F, Cochaux P, Beckman G, Samson M, Aksenova M, Cao A, et al. (Mar 1998). "The deltaccr5 mutation conferring protection against HIV-1 in Caucasian populations has a single and recent origin in Northeastern Europe". Human Molecular Genetics. 7 (3): 399–406. doi:10.1093/hmg/7.3.399. PMID 9466996.
- Lucotte G (2001). "Distribution of the CCR5 gene 32-basepair deletion in West Europe. A hypothesis about the possible dispersion of the mutation by the Vikings in historical times". Human Immunology. 62 (9): 933–6. doi:10.1016/S0198-8859(01)00292-0. PMID 11543895.
- Keele BF, Van Heuverswyn F, Li Y, Bailes E, Takehisa J, Santiago ML, Bibollet-Ruche F, Chen Y, Wain LV, Liegeois F, Loul S, Ngole EM, Bienvenue Y, Delaporte E, Brookfield JF, Sharp PM, Shaw GM, Peeters M, Hahn BH (Jul 2006). "Chimpanzee reservoirs of pandemic and nonpandemic HIV-1". Science. 313 (5786): 523–6. Bibcode:2006Sci...313..523K. doi:10.1126/science.1126531. PMC 2442710. PMID 16728595.
- Hahn BH, Shaw GM, De Cock KM, Sharp PM (Jan 2000). "AIDS as a zoonosis: scientific and public health implications". Science. 287 (5453): 607–14. Bibcode:2000Sci...287..607H. doi:10.1126/science.287.5453.607. PMID 10649986.
- Matic S. "HIV/AIDS in Europe: Moving from death sentence to chronic disease management" (PDF). World Health Organization.
- Faure E, Royer-Carenzi M (2008). "Is the European spatial distribution of the HIV-1-resistant CCR5-Δ32 allele formed by a breakdown of the pathocenosis due to the historical Roman expansion?". Infection, Genetics and Evolution. 8 (6): 864–874. doi:10.1016/j.meegid.2008.08.007. PMID 18790087.
- Dawson TC, Beck MA, Kuziel WA, Henderson F, Maeda N (2000). "Contrasting Effects of CCR5 and CCR2 Deficiency in the Pulmonary Inflammatory Response to Influenza A Virus". The American Journal of Pathology. 156 (6): 1951–1959. doi:10.1016/S0002-9440(10)65068-7. PMC 1850091. PMID 10854218.
- Escosteguy Vargas A, Cechim G, Ferraz Correa J, Gomes PA, de Souza Macedo G, de Medeiros RM, et al. (2009). "Pros and cons of a missing chemokine receptor—Comments on "Is the European spatial distribution of the HIV-1-resistant CCR5-Δ32 allele formed by a breakdown of the pathocenosis due to the historical Roman expansion?" by Eric Faure and Manuela Royer-Carenzi (2008)". Infection, Genetics and Evolution. 9 (4): 387–389. doi:10.1016/j.meegid.2009.01.001. PMID 19472441.
- O'Brien SJ, Dean M (Sep 1997). "In search of AIDS-resistance genes". Scientific American. 277 (3): 44–51. Bibcode:1997SciAm.277c..44O. doi:10.1038/scientificamerican0997-44. PMID 9274039.
- McEvedy C (Feb 1988). "The bubonic plague". Scientific American. 258 (2): 118–23. Bibcode:1988SciAm.258b.118M. doi:10.1038/scientificamerican0288-118. PMID 3055286.
- Galley C (1998). The Demography of Early Modern Towns: York in the Sixteenth and Seventeenth Centuries. London: MacMillan.
- Hatcher J (1977). Plague, Population, and the English Economy 1348-1530. London: MacMillan.
- Gottfried R (1983). The Black Death: Natural and Human Disaster in Medieval Europe. New York: Free Press.
- Keeling MJ, Gilligan CA (Nov 2000). "Bubonic plague: a metapopulation model of a zoonosis". Proceedings of the Royal Society B: Biological Sciences. 267 (1458): 2219–30. doi:10.1098/rspb.2000.1272. PMC 1690796. PMID 11413636.
- Appleby AB (1980). "The disappearance of plague: a continuing puzzle". The Economic History Review. 33 (2): 161–73. doi:10.2307/2595837. JSTOR 2595837. PMID 11614424.
- Russel J (1948). British Mediaeval Population. Albuquerque: University of New Mexico Press.
- Twigg G (1984). The Black Death: A Biological Reappraisal. London: Batsfod.
- Chapin C (1913). "Variation in the type of infectious diseases as shown by the history of smallpox in the United States, 1815-1912". J Infect Dis. 13 (2): 171–196. doi:10.1093/infdis/13.2.171.
- Fenner F (1998). Historical vignette: a life with poxviruses and publishers. Advances in Virus Research. 51. pp. 1–33. doi:10.1016/S0065-3527(08)60782-4. ISBN 9780120398515. PMID 9891584.
- Lalani AS, Masters J, Zeng W, Barrett J, Pannu R, Everett H, Arendt CW, McFadden G (Dec 1999). "Use of chemokine receptors by poxviruses". Science. 286 (5446): 1968–71. doi:10.1126/science.286.5446.1968. PMID 10583963.
- Novembre J, Galvani AP, Slatkin M (Nov 2005). "The geographic spread of the CCR5 Delta32 HIV-resistance allele". PLOS Biology. 3 (11): e339. doi:10.1371/journal.pbio.0030339. PMC 1255740. PMID 16216086.
- Klein RS (Jan 2008). "A moving target: the multiple roles of CCR5 in infectious diseases". The Journal of Infectious Diseases. 197 (2): 183–6. doi:10.1086/524692. PMID 18179384.
- Kindberg E, Mickiene A, Ax C, Akerlind B, Vene S, Lindquist L, Lundkvist A, Svensson L (Jan 2008). "A deletion in the chemokine receptor 5 (CCR5) gene is associated with tickborne encephalitis". The Journal of Infectious Diseases. 197 (2): 266–9. doi:10.1086/524709. PMID 18179389.
- Lim JK, Louie CY, Glaser C, Jean C, Johnson B, Johnson H, McDermott DH, Murphy PM (Jan 2008). "Genetic deficiency of chemokine receptor CCR5 is a strong risk factor for symptomatic West Nile virus infection: a meta-analysis of 4 cohorts in the US epidemic". The Journal of Infectious Diseases. 197 (2): 262–5. doi:10.1086/524691. PMID 18179388.
- Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM (Oct 2005). "Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection". The Journal of Experimental Medicine. 202 (8): 1087–98. doi:10.1084/jem.20042530. PMC 2213214. PMID 16230476.
- Steinberger P, Andris-Widhopf J, Bühler B, Torbett BE, Barbas CF (Jan 2000). "Functional deletion of the CCR5 receptor by intracellular immunization produces cells that are refractory to CCR5-dependent HIV-1 infection and cell fusion". Proceedings of the National Academy of Sciences of the United States of America. 97 (2): 805–10. Bibcode:2000PNAS...97..805S. doi:10.1073/pnas.97.2.805. PMC 15412. PMID 10639161.
- Schoofs M (2008-11-07). "A Doctor, a Mutation and a Potential Cure for AIDS". The Wall Street Journal. Retrieved 2010-12-15.
- Agrawal L, Lu X, Qingwen J, VanHorn-Ali Z, Nicolescu IV, McDermott DH, Murphy PM, Alkhatib G (Mar 2004). "Role for CCR5Delta32 protein in resistance to R5, R5X4, and X4 human immunodeficiency virus type 1 in primary CD4+ cells". Journal of Virology. 78 (5): 2277–87. doi:10.1128/JVI.78.5.2277-2287.2004. PMC 369216. PMID 14963124.
- "Autologous T-Cells Genetically Modified at the CCR5 Gene by Zinc Finger Nucleases SB-728 for HIV (Zinc-Finger)". U.S. National Institutes of Health. 2009-12-09. Retrieved 2009-12-30.
- Wade N (2009-12-28). "Zinc Fingers Could Be Key to Reviving Gene Therapy". The New York Times. Retrieved 2009-12-30.
- Corbyn Z (July 2012). "Plan launched to find HIV cure". Lancet. 380 (9838): 203–4. doi:10.1016/s0140-6736(12)61199-4. PMID 22826833.
- Gonzalez G, Park S, Chen D, Armitage S, Shpall E, Behringer R (September 2011). "Identification and frequency of CCR5Δ32/Δ32 HIV-resistant cord blood units from Houston area hospitals". HIV Medicine. 12 (8): 481–6. doi:10.1111/j.1468-1293.2010.00911.x. PMC 4021858. PMID 21375684.
- Petz LD, Burnett JC, Li H, Li S, Tonai R, Bakalinskaya M, Shpall EJ, Armitage S, Kurtzberg J, Regan DM, Clark P, Querol S, Gutman JA, Spellman SR, Gragert L, Rossi JJ (2015). "Progress toward curing HIV infection with hematopoietic cell transplantation". Stem Cells and Cloning: Advances and Applications. 8: 109–16. doi:10.2147/SCCAA.S56050. PMC 4524463. PMID 26251620.
- Begley S (28 November 2018). "Amid uproar, Chinese scientist defends creating gene-edited babies". STAT.
- Collins FS (2018-11-28). "Statement on Claim of First Gene-Edited Babies by Chinese Researcher". National Institutes of Health (NIH). U.S. Department of Health and Human Services.
Further reading
- Wilkinson D (Sep 1996). "Cofactors provide the entry keys. HIV-1". Current Biology. 6 (9): 1051–3. doi:10.1016/S0960-9822(02)70661-1. PMID 8805353.
- Broder CC, Dimitrov DS (1996). "HIV and the 7-transmembrane domain receptors". Pathobiology. 64 (4): 171–9. doi:10.1159/000164032. PMID 9031325.
- Choe H, Martin KA, Farzan M, Sodroski J, Gerard NP, Gerard C (Jun 1998). "Structural interactions between chemokine receptors, gp120 Env and CD4". Seminars in Immunology. 10 (3): 249–57. doi:10.1006/smim.1998.0127. PMID 9653051.
- Sheppard HW, Celum C, Michael NL, O'Brien S, Dean M, Carrington M, Dondero D, Buchbinder SP (Mar 2002). "HIV-1 infection in individuals with the CCR5-Delta32/Delta32 genotype: acquisition of syncytium-inducing virus at seroconversion". Journal of Acquired Immune Deficiency Syndromes. 29 (3): 307–13. doi:10.1097/00042560-200203010-00013. PMID 11873082.
- Freedman BD, Liu QH, Del Corno M, Collman RG (2003). "HIV-1 gp120 chemokine receptor-mediated signaling in human macrophages". Immunologic Research. 27 (2–3): 261–76. doi:10.1385/IR:27:2-3:261. PMID 12857973.
- Esté JA (Sep 2003). "Virus entry as a target for anti-HIV intervention". Current Medicinal Chemistry. 10 (17): 1617–32. doi:10.2174/0929867033457098. PMID 12871111.
- Gallo SA, Finnegan CM, Viard M, Raviv Y, Dimitrov A, Rawat SS, Puri A, Durell S, Blumenthal R (Jul 2003). "The HIV Env-mediated fusion reaction". Biochimica et Biophysica Acta (BBA) - Biomembranes. 1614 (1): 36–50. doi:10.1016/S0005-2736(03)00161-5. PMID 12873764.
- Zaitseva M, Peden K, Golding H (Jul 2003). "HIV coreceptors: role of structure, posttranslational modifications, and internalization in viral-cell fusion and as targets for entry inhibitors". Biochimica et Biophysica Acta (BBA) - Biomembranes. 1614 (1): 51–61. doi:10.1016/S0005-2736(03)00162-7. PMID 12873765.
- Lee C, Liu QH, Tomkowicz B, Yi Y, Freedman BD, Collman RG (Nov 2003). "Macrophage activation through CCR5- and CXCR4-mediated gp120-elicited signaling pathways". Journal of Leukocyte Biology. 74 (5): 676–82. doi:10.1189/jlb.0503206. PMID 12960231.
- Yi Y, Lee C, Liu QH, Freedman BD, Collman RG (2004). "Chemokine receptor utilization and macrophage signaling by human immunodeficiency virus type 1 gp120: Implications for neuropathogenesis". Journal of Neurovirology. 10 (Suppl 1): 91–6. doi:10.1080/753312758. PMID 14982745.
- Seibert C, Sakmar TP (2004). "Small-molecule antagonists of CCR5 and CXCR4: a promising new class of anti-HIV-1 drugs". Current Pharmaceutical Design. 10 (17): 2041–62. doi:10.2174/1381612043384312. PMID 15279544.
- Cutler CW, Jotwani R (2006). "Oral mucosal expression of HIV-1 receptors, co-receptors, and alpha-defensins: tableau of resistance or susceptibility to HIV infection?". Advances in Dental Research. 19 (1): 49–51. doi:10.1177/154407370601900110. PMC 3750741. PMID 16672549.
- Ajuebor MN, Carey JA, Swain MG (Aug 2006). "CCR5 in T cell-mediated liver diseases: what's going on?". Journal of Immunology. 177 (4): 2039–45. doi:10.4049/jimmunol.177.4.2039. PMID 16887960.
- Lipp M, Müller G (2003). "Shaping up adaptive immunity: the impact of CCR7 and CXCR5 on lymphocyte trafficking". Verhandlungen der Deutschen Gesellschaft für Pathologie. 87: 90–101. PMID 16888899.
- Balistreri CR, Caruso C, Grimaldi MP, Listì F, Vasto S, Orlando V, Campagna AM, Lio D, Candore G (Apr 2007). "CCR5 receptor: biologic and genetic implications in age-related diseases". Annals of the New York Academy of Sciences. 1100 (1): 162–72. Bibcode:2007NYASA1100..162B. doi:10.1196/annals.1395.014. PMID 17460174.
- Madsen HO, Poulsen K, Dahl O, Clark BF, Hjorth JP (Mar 1990). "Retropseudogenes constitute the major part of the human elongation factor 1 alpha gene family". Nucleic Acids Research. 18 (6): 1513–6. doi:10.1093/nar/18.6.1513. PMC 330519. PMID 2183196.
- Uetsuki T, Naito A, Nagata S, Kaziro Y (Apr 1989). "Isolation and characterization of the human chromosomal gene for polypeptide chain elongation factor-1 alpha". The Journal of Biological Chemistry. 264 (10): 5791–8. PMID 2564392.
- Whiteheart SW, Shenbagamurthi P, Chen L, Cotter RJ, Hart GW (Aug 1989). "Murine elongation factor 1 alpha (EF-1 alpha) is posttranslationally modified by novel amide-linked ethanolamine-phosphoglycerol moieties. Addition of ethanolamine-phosphoglycerol to specific glutamic acid residues on EF-1 alpha". The Journal of Biological Chemistry. 264 (24): 14334–41. PMID 2569467.
- Ann DK, Wu MM, Huang T, Carlson DM, Wu R (Mar 1988). "Retinol-regulated gene expression in human tracheobronchial epithelial cells. Enhanced expression of elongation factor EF-1 alpha". The Journal of Biological Chemistry. 263 (8): 3546–9. PMID 3346208.
- Brands JH, Maassen JA, van Hemert FJ, Amons R, Möller W (Feb 1986). "The primary structure of the alpha subunit of human elongation factor 1. Structural aspects of guanine-nucleotide-binding sites". European Journal of Biochemistry / FEBS. 155 (1): 167–71. doi:10.1111/j.1432-1033.1986.tb09472.x. PMID 3512269.
External links
- CCR5 - C-C motif chemokine receptor 5 (gene/pseudogene) - Homo sapiens (human) Information from NCBI Gene Database
- Crystal Structure of the CCR5 Chemokine Receptor A rotatable, zoomable 3-D image from rcsb.org
- Video and text from a PBS documentary about the discovery of CCR5
- "Chemokine Receptors: CCR5". IUPHAR Database of Receptors and Ion Channels. International Union of Basic and Clinical Pharmacology.
- HuGENavigator literature on HIV Infections and CCR5 from CDC - (note, authors may not be CDC employees, and there is no public domain notice on the page, so this cannot be assumed to be public domain)
- Schering-Plough Initiates Phase III Studies with CCR5-Vicriviroc in Treatment- Experienced HIV Patients.
- HIVcoPred A server for prediction of HIV coreceptor usage (CCR5). PLoS ONE 8(4): e61437
- Human CCR5 genome location and CCR5 gene details page in the UCSC Genome Browser.
- Overview of all the structural information available in the PDB for UniProt: P51681 (C-C chemokine receptor type 5) at the PDBe-KB.