Mitochondrion
The mitochondrion (/ˌmaɪtəˈkɒndrɪən/,[1] plural mitochondria) is a semi autonomous double-membrane-bound organelle found in most eukaryotic organisms. Some cells in some multicellular organisms may, however, lack mitochondria (for example, mature mammalian red blood cells). A number of unicellular organisms, such as microsporidia, parabasalids, and diplomonads, have also reduced or transformed their mitochondria into other structures.[2] To date, only one eukaryote, Monocercomonoides, is known to have completely lost its mitochondria,[3] and one multicellular organism, Henneguya salminicola, is known to have retained mitochondrion-related organelles in association with a complete loss of their mitochondrial genome.[3][4][5]

| Cell biology | |
|---|---|
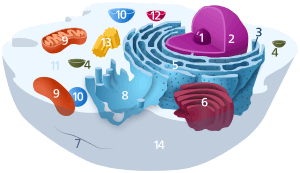 Components of a typical animal cell:
| |
 Components of a typical mitochondrion
1 Outer membrane
3 Lamella
4 Mitochondrial DNA |
The word mitochondrion comes from the Greek μίτος, mitos, "thread", and χονδρίον, chondrion, "granule"[6] or "grain-like". Mitochondria generate most of the cell's supply of adenosine triphosphate (ATP), used as a source of chemical energy.[7] A mitochondrion is thus termed the powerhouse of the cell.[8]
Mitochondria are commonly between 0.75 and 3 μm² in area[9] but vary considerably in size and structure. Unless specifically stained, they are not visible. In addition to supplying cellular energy, mitochondria are involved in other tasks, such as signaling, cellular differentiation, and cell death, as well as maintaining control of the cell cycle and cell growth.[10] Mitochondrial biogenesis is in turn temporally coordinated with these cellular processes.[11][12] Mitochondria have been implicated in several human diseases, such as mitochondrial disorders,[13] cardiac dysfunction,[14] heart failure[15] and autism.[16]
The number of mitochondria in a cell can vary widely by organism, tissue, and cell type. For instance, red blood cells have no mitochondria, whereas liver cells can have more than 2000.[17][18] The organelle is composed of compartments that carry out specialized functions. These compartments or regions include the outer membrane, the intermembrane space, the inner membrane, and the cristae and matrix.
Although most of a cell's DNA is contained in the cell nucleus, the mitochondrion has its own independent genome ("mitogenome") that shows substantial similarity to bacterial genomes.[19] Mitochondrial proteins (proteins transcribed from mitochondrial DNA) vary depending on the tissue and the species. In humans, 615 distinct types of proteins have been identified from cardiac mitochondria,[20] whereas in rats, 940 proteins have been reported.[21] The mitochondrial proteome is thought to be dynamically regulated.[22]
History of discovery and research
The first observations of intracellular structures that probably represented mitochondria were published in the 1840s.[23] Richard Altmann, in 1890, established them as cell organelles and called them "bioblasts".[23][24] The term "mitochondria" was coined by Carl Benda in 1898.[23][25] Leonor Michaelis discovered that Janus green can be used as a supravital stain for mitochondria in 1900. In 1904, Friedrich Meves, made the first recorded observation of mitochondria in plants in cells of the white waterlily, Nymphaea alba[23][26] and in 1908, along with Claudius Regaud, suggested that they contain proteins and lipids. Benjamin F. Kingsbury, in 1912, first related them with cell respiration, but almost exclusively based on morphological observations.[23] In 1913, particles from extracts of guinea-pig liver were linked to respiration by Otto Heinrich Warburg, which he called "grana". Warburg and Heinrich Otto Wieland, who had also postulated a similar particle mechanism, disagreed on the chemical nature of the respiration. It was not until 1925, when David Keilin discovered cytochromes, that the respiratory chain was described.[23]
In 1939, experiments using minced muscle cells demonstrated that cellular respiration using one oxygen atom can form two adenosine triphosphate (ATP) molecules, and, in 1941, the concept of the phosphate bonds of ATP being a form of energy in cellular metabolism was developed by Fritz Albert Lipmann. In the following years, the mechanism behind cellular respiration was further elaborated, although its link to the mitochondria was not known.[23] The introduction of tissue fractionation by Albert Claude allowed mitochondria to be isolated from other cell fractions and biochemical analysis to be conducted on them alone. In 1946, he concluded that cytochrome oxidase and other enzymes responsible for the respiratory chain were isolated to the mitochondria. Eugene Kennedy and Albert Lehninger discovered in 1948 that mitochondria are the site of oxidative phosphorylation in eukaryotes. Over time, the fractionation method was further developed, improving the quality of the mitochondria isolated, and other elements of cell respiration were determined to occur in the mitochondria.[23]
The first high-resolution electron micrographs appeared in 1952, replacing the Janus Green stains as the preferred way of visualizing the mitochondria.[23] This led to a more detailed analysis of the structure of the mitochondria, including confirmation that they were surrounded by a membrane. It also showed a second membrane inside the mitochondria that folded up in ridges dividing up the inner chamber and that the size and shape of the mitochondria varied from cell to cell.
The popular term "powerhouse of the cell" was coined by Philip Siekevitz in 1957.[8]
In 1967, it was discovered that mitochondria contained ribosomes.[27] In 1968, methods were developed for mapping the mitochondrial genes, with the genetic and physical map of yeast mitochondrial DNA being completed in 1976.[23]
Origin and evolution
There are two hypotheses about the origin of mitochondria: endosymbiotic and autogenous. The endosymbiotic hypothesis suggests that mitochondria were originally prokaryotic cells, capable of implementing oxidative mechanisms that were not possible for eukaryotic cells; they became endosymbionts living inside the eukaryote.[28] In the autogenous hypothesis, mitochondria were born by splitting off a portion of DNA from the nucleus of the eukaryotic cell at the time of divergence with the prokaryotes; this DNA portion would have been enclosed by membranes, which could not be crossed by proteins. Since mitochondria have many features in common with bacteria, the endosymbiotic hypothesis is more widely accepted.[28][29]
A mitochondrion contains DNA, which is organized as several copies of a single, usually circular chromosome. This mitochondrial chromosome contains genes for redox proteins, such as those of the respiratory chain. The CoRR hypothesis proposes that this co-location is required for redox regulation. The mitochondrial genome codes for some RNAs of ribosomes, and the 22 tRNAs necessary for the translation of mRNAs into protein. The circular structure is also found in prokaryotes. The proto-mitochondrion was probably closely related to Rickettsia.[30][31] However, the exact relationship of the ancestor of mitochondria to the alphaproteobacteria and whether the mitochondrion was formed at the same time or after the nucleus, remains controversial.[32] For example, it has been suggested that the SAR11 clade of bacteria shares a relatively recent common ancestor with the mitochondria,[33] while phylogenomic analyses indicate that mitochondria evolved from a proteobacteria lineage that is closely related to or a member of alphaproteobacteria.[34][35]
| Schematic ribosomal RNA phylogeny of Alphaproteobacteria | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| The cladogram of Rickettsidae has been inferred by Ferla et al. [36] from the comparison of 16S + 23S ribosomal RNA sequences. |
The ribosomes coded for by the mitochondrial DNA are similar to those from bacteria in size and structure.[37] They closely resemble the bacterial 70S ribosome and not the 80S cytoplasmic ribosomes, which are coded for by nuclear DNA.
The endosymbiotic relationship of mitochondria with their host cells was popularized by Lynn Margulis.[38] The endosymbiotic hypothesis suggests that mitochondria descended from bacteria that somehow survived endocytosis by another cell, and became incorporated into the cytoplasm. The ability of these bacteria to conduct respiration in host cells that had relied on glycolysis and fermentation would have provided a considerable evolutionary advantage. This symbiotic relationship probably developed 1.7 to 2 billion years ago.[39][40] A few groups of unicellular eukaryotes have only vestigial mitochondria or derived structures: the microsporidians, metamonads, and archamoebae.[41] These groups appear as the most primitive eukaryotes on phylogenetic trees constructed using rRNA information, which once suggested that they appeared before the origin of mitochondria. However, this is now known to be an artifact of long-branch attraction—they are derived groups and retain genes or organelles derived from mitochondria (e. g., mitosomes and hydrogenosomes).[2] By this, mitochondria, hydrogenosomes, mitosomes, and related organelles as found in some loricifera (e. g. Spinoloricus)[42][43] and myxozoa (e. g. Henneguya zschokkei) are together classified as MROs, mitochondrion-related organelles.[44][45]
Monocercomonoides appear to have lost their mitochondria completely and at least some of the mitochondrial functions seem to be carried out by cytoplasmic proteins now.[46]
Structure

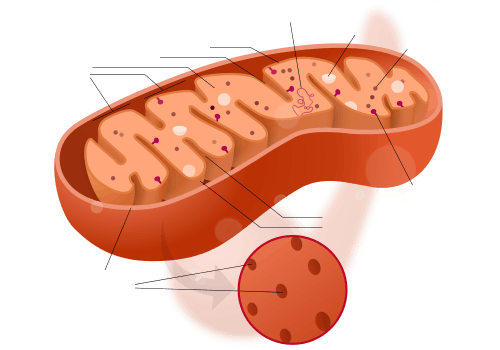
A mitochondrion contains outer and inner membranes composed of phospholipid bilayers and proteins.[17] The two membranes have different properties. Because of this double-membraned organization, there are five distinct parts to a mitochondrion. They are:
- the outer mitochondrial membrane,
- the intermembrane space (the space between the outer and inner membranes),
- the inner mitochondrial membrane,
- the cristae space (formed by infoldings of the inner membrane), and
- the matrix (space within the inner membrane).
Mitochondria stripped of their outer membrane are called mitoplasts.
Outer membrane
The outer mitochondrial membrane, which encloses the entire organelle, is 60 to 75 angstroms (Å) thick. It has a protein-to-phospholipid ratio similar to that of the cell membrane (about 1:1 by weight). It contains large numbers of integral membrane proteins called porins. A major trafficking protein is the pore-forming voltage-dependent anion channel (VDAC). The VDAC is the primary transporter of nucleotides, ions and metabolites between the cytosol and the intermembrane space.[48][49] It is formed as a beta barrel that spans the outer membrane, similar to that in the gram-negative bacterial membrane.[50] Larger proteins can enter the mitochondrion if a signaling sequence at their N-terminus binds to a large multisubunit protein called translocase in the outer membrane, which then actively moves them across the membrane.[51] Mitochondrial pro-proteins are imported through specialised translocation complexes.
The outer membrane also contains enzymes involved in such diverse activities as the elongation of fatty acids, oxidation of epinephrine, and the degradation of tryptophan. These enzymes include monoamine oxidase, rotenone-insensitive NADH-cytochrome c-reductase, kynurenine hydroxylase and fatty acid Co-A ligase. Disruption of the outer membrane permits proteins in the intermembrane space to leak into the cytosol, leading to certain cell death.[52] The mitochondrial outer membrane can associate with the endoplasmic reticulum (ER) membrane, in a structure called MAM (mitochondria-associated ER-membrane). This is important in the ER-mitochondria calcium signaling and is involved in the transfer of lipids between the ER and mitochondria.[53] Outside the outer membrane there are small (diameter: 60Å) particles named sub-units of Parson.
Intermembrane space
The mitochondrial intermembrane space is the space between the outer membrane and the inner membrane. It is also known as perimitochondrial space. Because the outer membrane is freely permeable to small molecules, the concentrations of small molecules, such as ions and sugars, in the intermembrane space is the same as in the cytosol.[17] However, large proteins must have a specific signaling sequence to be transported across the outer membrane, so the protein composition of this space is different from the protein composition of the cytosol. One protein that is localized to the intermembrane space in this way is cytochrome c.[52]
Inner membrane
The inner mitochondrial membrane contains proteins with three types of functions:[17]
- Those that perform the electron transport chain redox reactions
- ATP synthase, which generates ATP in the matrix
- Specific transport proteins that regulate metabolite passage into and out of the mitochondrial matrix
It contains more than 151 different polypeptides, and has a very high protein-to-phospholipid ratio (more than 3:1 by weight, which is about 1 protein for 15 phospholipids). The inner membrane is home to around 1/5 of the total protein in a mitochondrion.[54] Additionally, the inner membrane is rich in an unusual phospholipid, cardiolipin. This phospholipid was originally discovered in cow hearts in 1942, and is usually characteristic of mitochondrial and bacterial plasma membranes.[55] Cardiolipin contains four fatty acids rather than two, and may help to make the inner membrane impermeable.[17] Unlike the outer membrane, the inner membrane does not contain porins, and is highly impermeable to all molecules. Almost all ions and molecules require special membrane transporters to enter or exit the matrix. Proteins are ferried into the matrix via the translocase of the inner membrane (TIM) complex or via Oxa1.[51] In addition, there is a membrane potential across the inner membrane, formed by the action of the enzymes of the electron transport chain. Inner membrane fusion is mediated by the inner membrane protein OPA1.[56]
Cristae

The inner mitochondrial membrane is compartmentalized into numerous cristae, which expand the surface area of the inner mitochondrial membrane, enhancing its ability to produce ATP. For typical liver mitochondria, the area of the inner membrane is about five times as large as the outer membrane. This ratio is variable and mitochondria from cells that have a greater demand for ATP, such as muscle cells, contain even more cristae. Mitochondria within the same cell can have substantially different crista-density, the ones that are required to produce more energy, have much more crista-membrane surface.[57] These folds are studded with small round bodies known as F1 particles or oxysomes. These are not simple random folds but rather invaginations of the inner membrane, which can affect overall chemiosmotic function.[58]
One recent mathematical modeling study has suggested that the optical properties of the cristae in filamentous mitochondria may affect the generation and propagation of light within the tissue.[59]
Matrix
The matrix is the space enclosed by the inner membrane. It contains about 2/3 of the total proteins in a mitochondrion.[17] The matrix is important in the production of ATP with the aid of the ATP synthase contained in the inner membrane. The matrix contains a highly concentrated mixture of hundreds of enzymes, special mitochondrial ribosomes, tRNA, and several copies of the mitochondrial DNA genome. Of the enzymes, the major functions include oxidation of pyruvate and fatty acids, and the citric acid cycle.[17] The DNA molecules are packaged into nucleoids by proteins, one of which is TFAM.[60]
Mitochondria have their own genetic material, and the machinery to manufacture their own RNAs and proteins (see: protein biosynthesis). A published human mitochondrial DNA sequence revealed 16,569 base pairs encoding 37 genes: 22 tRNA, 2 rRNA, and 13 peptide genes.[61] The 13 mitochondrial peptides in humans are integrated into the inner mitochondrial membrane, along with proteins encoded by genes that reside in the host cell's nucleus.
Mitochondria-associated ER membrane (MAM)
The mitochondria-associated ER membrane (MAM) is another structural element that is increasingly recognized for its critical role in cellular physiology and homeostasis. Once considered a technical snag in cell fractionation techniques, the alleged ER vesicle contaminants that invariably appeared in the mitochondrial fraction have been re-identified as membranous structures derived from the MAM—the interface between mitochondria and the ER.[62] Physical coupling between these two organelles had previously been observed in electron micrographs and has more recently been probed with fluorescence microscopy.[62] Such studies estimate that at the MAM, which may comprise up to 20% of the mitochondrial outer membrane, the ER and mitochondria are separated by a mere 10–25 nm and held together by protein tethering complexes.[62][53][63]
Purified MAM from subcellular fractionation has been shown to be enriched in enzymes involved in phospholipid exchange, in addition to channels associated with Ca2+ signaling.[62][63] These hints of a prominent role for the MAM in the regulation of cellular lipid stores and signal transduction have been borne out, with significant implications for mitochondrial-associated cellular phenomena, as discussed below. Not only has the MAM provided insight into the mechanistic basis underlying such physiological processes as intrinsic apoptosis and the propagation of calcium signaling, but it also favors a more refined view of the mitochondria. Though often seen as static, isolated 'powerhouses' hijacked for cellular metabolism through an ancient endosymbiotic event, the evolution of the MAM underscores the extent to which mitochondria have been integrated into overall cellular physiology, with intimate physical and functional coupling to the endomembrane system.
Phospholipid transfer
The MAM is enriched in enzymes involved in lipid biosynthesis, such as phosphatidylserine synthase on the ER face and phosphatidylserine decarboxylase on the mitochondrial face.[64][65] Because mitochondria are dynamic organelles constantly undergoing fission and fusion events, they require a constant and well-regulated supply of phospholipids for membrane integrity.[66][67] But mitochondria are not only a destination for the phospholipids they finish synthesis of; rather, this organelle also plays a role in inter-organelle trafficking of the intermediates and products of phospholipid biosynthetic pathways, ceramide and cholesterol metabolism, and glycosphingolipid anabolism.[65][67]
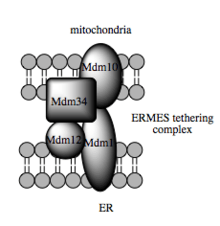
Such trafficking capacity depends on the MAM, which has been shown to facilitate transfer of lipid intermediates between organelles.[64] In contrast to the standard vesicular mechanism of lipid transfer, evidence indicates that the physical proximity of the ER and mitochondrial membranes at the MAM allows for lipid flipping between opposed bilayers.[67] Despite this unusual and seemingly energetically unfavorable mechanism, such transport does not require ATP.[67] Instead, in yeast, it has been shown to be dependent on a multiprotein tethering structure termed the ER-mitochondria encounter structure, or ERMES, although it remains unclear whether this structure directly mediates lipid transfer or is required to keep the membranes in sufficiently close proximity to lower the energy barrier for lipid flipping.[67][68]
The MAM may also be part of the secretory pathway, in addition to its role in intracellular lipid trafficking. In particular, the MAM appears to be an intermediate destination between the rough ER and the Golgi in the pathway that leads to very-low-density lipoprotein, or VLDL, assembly and secretion.[65][69] The MAM thus serves as a critical metabolic and trafficking hub in lipid metabolism.
Calcium signaling
A critical role for the ER in calcium signaling was acknowledged before such a role for the mitochondria was widely accepted, in part because the low affinity of Ca2+ channels localized to the outer mitochondrial membrane seemed to contradict this organelle's purported responsiveness to changes in intracellular Ca2+ flux.[62][70] But the presence of the MAM resolves this apparent contradiction: the close physical association between the two organelles results in Ca2+ microdomains at contact points that facilitate efficient Ca2+ transmission from the ER to the mitochondria.[62] Transmission occurs in response to so-called "Ca2+ puffs" generated by spontaneous clustering and activation of IP3R, a canonical ER membrane Ca2+ channel.[62][53]
The fate of these puffs—in particular, whether they remain restricted to isolated locales or integrated into Ca2+ waves for propagation throughout the cell—is determined in large part by MAM dynamics. Although reuptake of Ca2+ by the ER (concomitant with its release) modulates the intensity of the puffs, thus insulating mitochondria to a certain degree from high Ca2+ exposure, the MAM often serves as a firewall that essentially buffers Ca2+ puffs by acting as a sink into which free ions released into the cytosol can be funneled.[62][71][72] This Ca2+ tunneling occurs through the low-affinity Ca2+ receptor VDAC1, which recently has been shown to be physically tethered to the IP3R clusters on the ER membrane and enriched at the MAM.[62][53][73] The ability of mitochondria to serve as a Ca2+ sink is a result of the electrochemical gradient generated during oxidative phosphorylation, which makes tunneling of the cation an exergonic process.[73] Normal, mild calcium influx from cytosol into the mitochondrial matrix causes transient depolarization that is corrected by pumping out protons.
But transmission of Ca2+ is not unidirectional; rather, it is a two-way street.[70] The properties of the Ca2+ pump SERCA and the channel IP3R present on the ER membrane facilitate feedback regulation coordinated by MAM function. In particular, the clearance of Ca2+ by the MAM allows for spatio-temporal patterning of Ca2+ signaling because Ca2+ alters IP3R activity in a biphasic manner.[62] SERCA is likewise affected by mitochondrial feedback: uptake of Ca2+ by the MAM stimulates ATP production, thus providing energy that enables SERCA to reload the ER with Ca2+ for continued Ca2+ efflux at the MAM.[71][73] Thus, the MAM is not a passive buffer for Ca2+ puffs; rather it helps modulate further Ca2+ signaling through feedback loops that affect ER dynamics.
Regulating ER release of Ca2+ at the MAM is especially critical because only a certain window of Ca2+ uptake sustains the mitochondria, and consequently the cell, at homeostasis. Sufficient intraorganelle Ca2+ signaling is required to stimulate metabolism by activating dehydrogenase enzymes critical to flux through the citric acid cycle.[74] However, once Ca2+ signaling in the mitochondria passes a certain threshold, it stimulates the intrinsic pathway of apoptosis in part by collapsing the mitochondrial membrane potential required for metabolism.[62] Studies examining the role of pro- and anti-apoptotic factors support this model; for example, the anti-apoptotic factor Bcl-2 has been shown to interact with IP3Rs to reduce Ca2+ filling of the ER, leading to reduced efflux at the MAM and preventing collapse of the mitochondrial membrane potential post-apoptotic stimuli.[62] Given the need for such fine regulation of Ca2+ signaling, it is perhaps unsurprising that dysregulated mitochondrial Ca2+ has been implicated in several neurodegenerative diseases, while the catalogue of tumor suppressors includes a few that are enriched at the MAM.[73]
Molecular basis for tethering
Recent advances in the identification of the tethers between the mitochondrial and ER membranes suggest that the scaffolding function of the molecular elements involved is secondary to other, non-structural functions. In yeast, ERMES, a multiprotein complex of interacting ER- and mitochondrial-resident membrane proteins, is required for lipid transfer at the MAM and exemplifies this principle. One of its components, for example, is also a constituent of the protein complex required for insertion of transmembrane beta-barrel proteins into the lipid bilayer.[67] However, a homologue of the ERMES complex has not yet been identified in mammalian cells. Other proteins implicated in scaffolding likewise have functions independent of structural tethering at the MAM; for example, ER-resident and mitochondrial-resident mitofusins form heterocomplexes that regulate the number of inter-organelle contact sites, although mitofusins were first identified for their role in fission and fusion events between individual mitochondria.[62] Glucose-related protein 75 (grp75) is another dual-function protein. In addition to the matrix pool of grp75, a portion serves as a chaperone that physically links the mitochondrial and ER Ca2+ channels VDAC and IP3R for efficient Ca2+ transmission at the MAM.[62][53] Another potential tether is Sigma-1R, a non-opioid receptor whose stabilization of ER-resident IP3R may preserve communication at the MAM during the metabolic stress response.[75][76]
Perspective
The MAM is a critical signaling, metabolic, and trafficking hub in the cell that allows for the integration of ER and mitochondrial physiology. Coupling between these organelles is not simply structural but functional as well and critical for overall cellular physiology and homeostasis. The MAM thus offers a perspective on mitochondria that diverges from the traditional view of this organelle as a static, isolated unit appropriated for its metabolic capacity by the cell.[77] Instead, this mitochondrial-ER interface emphasizes the integration of the mitochondria, the product of an endosymbiotic event, into diverse cellular processes. Recently it has also been shown, that mitochondria and MAM-s in neurons are anchored to specialised intercellular communication sites (so called somatic-junctions). Microglial processes monitor and protect neuronal functions at these sites, and MAM-s are supposed to have an important role in this type of cellular quality-control.[78]
Organization and distribution
Mitochondria (and related structures) are found in all eukaryotes (except two—the Oxymonad Monocercomonoides and Henneguya salminicola).[3][3][79][5][80] Although commonly depicted as bean-like structures they form a highly dynamic network in the majority of cells where they constantly undergo fission and fusion. The population of all the mitochondria of a given cell constitutes the chondriome.[81] Mitochondria vary in number and location according to cell type. A single mitochondrion is often found in unicellular organisms. Conversely, the chondriome size of human liver cells is large, with about 1000–2000 mitochondria per cell, making up 1/5 of the cell volume.[17] The mitochondrial content of otherwise similar cells can vary substantially in size and membrane potential,[82] with differences arising from sources including uneven partitioning at cell divisions, leading to extrinsic differences in ATP levels and downstream cellular processes.[83] The mitochondria can be found nestled between myofibrils of muscle or wrapped around the sperm flagellum.[17] Often, they form a complex 3D branching network inside the cell with the cytoskeleton. The association with the cytoskeleton determines mitochondrial shape, which can affect the function as well:[84] different structures of the mitochondrial network may afford the population a variety of physical, chemical, and signalling advantages or disadvantages.[85] Mitochondria in cells are always distributed along microtubules and the distribution of these organelles is also correlated with the endoplasmic reticulum.[86] Recent evidence suggests that vimentin, one of the components of the cytoskeleton, is also critical to the association with the cytoskeleton.[87]
Function
The most prominent roles of mitochondria are to produce the energy currency of the cell, ATP (i.e., phosphorylation of ADP), through respiration, and to regulate cellular metabolism.[18] The central set of reactions involved in ATP production are collectively known as the citric acid cycle, or the Krebs cycle. However, the mitochondrion has many other functions in addition to the production of ATP.
Energy conversion
A dominant role for the mitochondria is the production of ATP, as reflected by the large number of proteins in the inner membrane for this task. This is done by oxidizing the major products of glucose: pyruvate, and NADH, which are produced in the cytosol.[18] This type of cellular respiration known as aerobic respiration, is dependent on the presence of oxygen, which provides most of the energy released.[88] When oxygen is limited, the glycolytic products will be metabolized by anaerobic fermentation, a process that is independent of the mitochondria.[18] The production of ATP from glucose and oxygen has an approximately 13-times higher yield during aerobic respiration compared to fermentation.[89] Plant mitochondria can also produce a limited amount of ATP without oxygen by using the alternate substrate nitrite.[90] ATP crosses out through the inner membrane with the help of a specific protein, and across the outer membrane via porins. ADP returns via the same route.
Pyruvate and the citric acid cycle
Pyruvate molecules produced by glycolysis are actively transported across the inner mitochondrial membrane, and into the matrix where they can either be oxidized and combined with coenzyme A to form CO2, acetyl-CoA, and NADH,[18] or they can be carboxylated (by pyruvate carboxylase) to form oxaloacetate. This latter reaction ”fills up” the amount of oxaloacetate in the citric acid cycle, and is therefore an anaplerotic reaction, increasing the cycle's capacity to metabolize acetyl-CoA when the tissue's energy needs (e.g. in muscle) are suddenly increased by activity.[91]
In the citric acid cycle, all the intermediates (e.g. citrate, iso-citrate, alpha-ketoglutarate, succinate, fumarate, malate and oxaloacetate) are regenerated during each turn of the cycle. Adding more of any of these intermediates to the mitochondrion therefore means that the additional amount is retained within the cycle, increasing all the other intermediates as one is converted into the other. Hence, the addition of any one of them to the cycle has an anaplerotic effect, and its removal has a cataplerotic effect. These anaplerotic and cataplerotic reactions will, during the course of the cycle, increase or decrease the amount of oxaloacetate available to combine with acetyl-CoA to form citric acid. This in turn increases or decreases the rate of ATP production by the mitochondrion, and thus the availability of ATP to the cell.[91]
Acetyl-CoA, on the other hand, derived from pyruvate oxidation, or from the beta-oxidation of fatty acids, is the only fuel to enter the citric acid cycle. With each turn of the cycle one molecule of acetyl-CoA is consumed for every molecule of oxaloacetate present in the mitochondrial matrix, and is never regenerated. It is the oxidation of the acetate portion of acetyl-CoA that produces CO2 and water, with the energy thus released captured in the form of ATP.[91]
In the liver, the carboxylation of cytosolic pyruvate into intra-mitochondrial oxaloacetate is an early step in the gluconeogenic pathway, which converts lactate and de-aminated alanine into glucose,[18][91] under the influence of high levels of glucagon and/or epinephrine in the blood.[91] Here, the addition of oxaloacetate to the mitochondrion does not have a net anaplerotic effect, as another citric acid cycle intermediate (malate) is immediately removed from the mitochondrion to be converted into cytosolic oxaloacetate, which is ultimately converted into glucose, in a process that is almost the reverse of glycolysis.[91]
The enzymes of the citric acid cycle are located in the mitochondrial matrix, with the exception of succinate dehydrogenase, which is bound to the inner mitochondrial membrane as part of Complex II.[92] The citric acid cycle oxidizes the acetyl-CoA to carbon dioxide, and, in the process, produces reduced cofactors (three molecules of NADH and one molecule of FADH2) that are a source of electrons for the electron transport chain, and a molecule of GTP (that is readily converted to an ATP).[18]
NADH and FADH2: the electron transport chain
The electrons from NADH and FADH2 are transferred to oxygen (O2), an energy-rich molecule,[88] and hydrogen (protons) in several steps via the electron transport chain. NADH and FADH2 molecules are produced within the matrix via the citric acid cycle but are also produced in the cytoplasm by glycolysis. Reducing equivalents from the cytoplasm can be imported via the malate-aspartate shuttle system of antiporter proteins or feed into the electron transport chain using a glycerol phosphate shuttle.[18] Protein complexes in the inner membrane (NADH dehydrogenase (ubiquinone), cytochrome c reductase, and cytochrome c oxidase) perform the transfer and the incremental release of energy is used to pump protons (H+) into the intermembrane space. This process is efficient, but a small percentage of electrons may prematurely reduce oxygen, forming reactive oxygen species such as superoxide.[18] This can cause oxidative stress in the mitochondria and may contribute to the decline in mitochondrial function associated with the aging process.[93]
As the proton concentration increases in the intermembrane space, a strong electrochemical gradient is established across the inner membrane. The protons can return to the matrix through the ATP synthase complex, and their potential energy is used to synthesize ATP from ADP and inorganic phosphate (Pi).[18] This process is called chemiosmosis, and was first described by Peter Mitchell,[94][95] who was awarded the 1978 Nobel Prize in Chemistry for his work. Later, part of the 1997 Nobel Prize in Chemistry was awarded to Paul D. Boyer and John E. Walker for their clarification of the working mechanism of ATP synthase.[96]
Heat production
Under certain conditions, protons can re-enter the mitochondrial matrix without contributing to ATP synthesis. This process is known as proton leak or mitochondrial uncoupling and is due to the facilitated diffusion of protons into the matrix. The process results in the unharnessed potential energy of the proton electrochemical gradient being released as heat.[18] The process is mediated by a proton channel called thermogenin, or UCP1.[97] Thermogenin is a 33 kDa protein first discovered in 1973.[98] Thermogenin is primarily found in brown adipose tissue, or brown fat, and is responsible for non-shivering thermogenesis. Brown adipose tissue is found in mammals, and is at its highest levels in early life and in hibernating animals. In humans, brown adipose tissue is present at birth and decreases with age.[97]
Storage of calcium ions
The concentrations of free calcium in the cell can regulate an array of reactions and is important for signal transduction in the cell. Mitochondria can transiently store calcium, a contributing process for the cell's homeostasis of calcium.[99] [100] In fact, their ability to rapidly take in calcium for later release makes them very good "cytosolic buffers" for calcium.[101][102][103] The endoplasmic reticulum (ER) is the most significant storage site of calcium,[70] and there is a significant interplay between the mitochondrion and ER with regard to calcium.[104] The calcium is taken up into the matrix by the mitochondrial calcium uniporter on the inner mitochondrial membrane.[105] It is primarily driven by the mitochondrial membrane potential.[100] Release of this calcium back into the cell's interior can occur via a sodium-calcium exchange protein or via "calcium-induced-calcium-release" pathways.[105] This can initiate calcium spikes or calcium waves with large changes in the membrane potential. These can activate a series of second messenger system proteins that can coordinate processes such as neurotransmitter release in nerve cells and release of hormones in endocrine cells.[106]
Ca2+ influx to the mitochondrial matrix has recently been implicated as a mechanism to regulate respiratory bioenergetics by allowing the electrochemical potential across the membrane to transiently "pulse" from ΔΨ-dominated to pH-dominated, facilitating a reduction of oxidative stress.[107] In neurons, concomitant increases in cytosolic and mitochondrial calcium act to synchronize neuronal activity with mitochondrial energy metabolism. Mitochondrial matrix calcium levels can reach the tens of micromolar levels, which is necessary for the activation of isocitrate dehydrogenase, one of the key regulatory enzymes of the Krebs cycle.[108]
Additional functions
Mitochondria play a central role in many other metabolic tasks, such as:
- Signaling through mitochondrial reactive oxygen species[109]
- Regulation of the membrane potential[18]
- Apoptosis-programmed cell death[110]
- Calcium signaling (including calcium-evoked apoptosis)[111]
- Regulation of cellular metabolism[10]
- Certain heme synthesis reactions[112] (see also: porphyrin)
- Steroid synthesis.[101]
- Hormonal signaling [113] Mitochondria are sensitive and responsive to hormones, in part by the action of mitochondrial estrogen receptors (mtERs). These receptors have been found in various tissues and cell types, including brain [114] and heart [115]
- Immune signaling [116]
- Neuronal mitochondria also contribute to cellular quality control by reporting neuronal status towards microglia through specialised somatic-junctions.[78]
Some mitochondrial functions are performed only in specific types of cells. For example, mitochondria in liver cells contain enzymes that allow them to detoxify ammonia, a waste product of protein metabolism. A mutation in the genes regulating any of these functions can result in mitochondrial diseases.
Cellular proliferation regulation
The relationship between cellular proliferation and mitochondria has been investigated using cervical cancer HeLa cells. Tumor cells require an ample amount of ATP (Adenosine triphosphate) in order to synthesize bioactive compounds such as lipids, proteins, and nucleotides for rapid cell proliferation.[117] The majority of ATP in tumor cells is generated via the oxidative phosphorylation pathway (OxPhos).[118] Interference with OxPhos have shown to cause cell cycle arrest suggesting that mitochondria play a role in cell proliferation.[118] Mitochondrial ATP production is also vital for cell division and differentiation in infection [119] in addition to basic functions in the cell including the regulation of cell volume, solute concentration, and cellular architecture.[120][121][122] ATP levels differ at various stages of the cell cycle suggesting that there is a relationship between the abundance of ATP and the cell's ability to enter a new cell cycle.[123] ATP's role in the basic functions of the cell make the cell cycle sensitive to changes in the availability of mitochondrial derived ATP.[123] The variation in ATP levels at different stages of the cell cycle support the hypothesis that mitochondria play an important role in cell cycle regulation.[123] Although the specific mechanisms between mitochondria and the cell cycle regulation is not well understood, studies have shown that low energy cell cycle checkpoints monitor the energy capability before committing to another round of cell division.[10]
Genome

Mitochondria contain their own genome, an indication that they are derived from bacteria through endosymbiosis. However, the ancestral endosymbiont genome has lost most of its genes so that the mitochondrial genome (mitogenome) is one of the most reduced genomes across organisms.
The human mitochondrial genome is a circular DNA molecule of about 16 kilobases.[124] It encodes 37 genes: 13 for subunits of respiratory complexes I, III, IV and V, 22 for mitochondrial tRNA (for the 20 standard amino acids, plus an extra gene for leucine and serine), and 2 for rRNA.[124] One mitochondrion can contain two to ten copies of its DNA.[125]
As in prokaryotes, there is a very high proportion of coding DNA and an absence of repeats. Mitochondrial genes are transcribed as multigenic transcripts, which are cleaved and polyadenylated to yield mature mRNAs. Not all proteins necessary for mitochondrial function are encoded by the mitochondrial genome; most are coded by genes in the cell nucleus and the corresponding proteins are imported into the mitochondrion.[61] The exact number of genes encoded by the nucleus and the mitochondrial genome differs between species. Most mitochondrial genomes are circular, although exceptions have been reported.[126] In general, mitochondrial DNA lacks introns, as is the case in the human mitochondrial genome;[61] however, introns have been observed in some eukaryotic mitochondrial DNA,[127] such as that of yeast[128] and protists,[129] including Dictyostelium discoideum.[130] Between protein-coding regions, tRNAs are present. During transcription, the tRNAs acquire their characteristic L-shape that gets recognized and cleaved by specific enzymes. Mitochondrial tRNA genes have different sequences from the nuclear tRNAs but lookalikes of mitochondrial tRNAs have been found in the nuclear chromosomes with high sequence similarity.[131]
In animals, the mitochondrial genome is typically a single circular chromosome that is approximately 16 kb long and has 37 genes. The genes, while highly conserved, may vary in location. Curiously, this pattern is not found in the human body louse (Pediculus humanus). Instead, this mitochondrial genome is arranged in 18 minicircular chromosomes, each of which is 3–4 kb long and has one to three genes.[132] This pattern is also found in other sucking lice, but not in chewing lice. Recombination has been shown to occur between the minichromosomes. The reason for this difference is not known.
Alternative genetic code
While slight variations on the standard genetic code had been predicted earlier,[133] none was discovered until 1979, when researchers studying human mitochondrial genes determined that they used an alternative code.[134] However, the mitochondria of many other eukaryotes, including most plants, use the standard code.[135] Many slight variants have been discovered since,[136] including various alternative mitochondrial codes.[137] Further, the AUA, AUC, and AUU codons are all allowable start codons.
| Organism | Codon | Standard | Mitochondria |
|---|---|---|---|
| Mammals | AGA, AGG | Arginine | Stop codon |
| Invertebrates | AGA, AGG | Arginine | Serine |
| Fungi | CUA | Leucine | Threonine |
| All of the above | AUA | Isoleucine | Methionine |
| UGA | Stop codon | Tryptophan |
Some of these differences should be regarded as pseudo-changes in the genetic code due to the phenomenon of RNA editing, which is common in mitochondria. In higher plants, it was thought that CGG encoded for tryptophan and not arginine; however, the codon in the processed RNA was discovered to be the UGG codon, consistent with the standard genetic code for tryptophan.[138] Of note, the arthropod mitochondrial genetic code has undergone parallel evolution within a phylum, with some organisms uniquely translating AGG to lysine.[139]
Evolution and diversity
Mitochondrial genomes have far fewer genes than the bacteria from which they are thought to be descended. Although some have been lost altogether, many have been transferred to the nucleus, such as the respiratory complex II protein subunits.[124] This is thought to be relatively common over evolutionary time. A few organisms, such as the Cryptosporidium, actually have mitochondria that lack any DNA, presumably because all their genes have been lost or transferred.[140] In Cryptosporidium, the mitochondria have an altered ATP generation system that renders the parasite resistant to many classical mitochondrial inhibitors such as cyanide, azide, and atovaquone.[140]
Replication and inheritance
Mitochondria divide by binary fission, similar to bacterial cell division.[141] The regulation of this division differs between eukaryotes. In many single-celled eukaryotes, their growth and division are linked to the cell cycle. For example, a single mitochondrion may divide synchronously with the nucleus. This division and segregation process must be tightly controlled so that each daughter cell receives at least one mitochondrion. In other eukaryotes (in mammals for example), mitochondria may replicate their DNA and divide mainly in response to the energy needs of the cell, rather than in phase with the cell cycle. When the energy needs of a cell are high, mitochondria grow and divide. When energy use is low, mitochondria are destroyed or become inactive. In such examples, and in contrast to the situation in many single-celled eukaryotes, mitochondria are apparently randomly distributed to the daughter cells during the division of the cytoplasm. Understanding of mitochondrial dynamics, which is described as the balance between mitochondrial fusion and fission, has revealed that functional and structural alterations in mitochondrial morphology are important factors in pathologies associated with several disease conditions.[142]
The hypothesis of mitochondrial binary fission has relied on the visualization by fluorescence microscopy and conventional transmission electron microscopy (TEM). The resolution of fluorescence microscopy(~200 nm) is insufficient to distinguish structural details, such as double mitochondrial membrane in mitochondrial division or even to distinguish individual mitochondria when several are close together. Conventional TEM has also some technical limitations in verifying mitochondrial division. Cryo-electron tomography was recently used to visualize mitochondrial division in frozen hydrated intact cells. It revealed that mitochondria divide by budding.[143]
An individual's mitochondrial genes are not inherited by the same mechanism as nuclear genes. Typically, the mitochondria are inherited from one parent only. In humans, when an egg cell is fertilized by a sperm, the egg nucleus and sperm nucleus each contribute equally to the genetic makeup of the zygote nucleus. In contrast, the mitochondria, and therefore the mitochondrial DNA, usually come from the egg only. The sperm's mitochondria enter the egg, but do not contribute genetic information to the embryo.[144] Instead, paternal mitochondria are marked with ubiquitin to select them for later destruction inside the embryo.[145] The egg cell contains relatively few mitochondria, but it is these mitochondria that survive and divide to populate the cells of the adult organism. Mitochondria are, therefore, in most cases inherited only from mothers, a pattern known as maternal inheritance. This mode is seen in most organisms, including the majority of animals. However, mitochondria in some species can sometimes be inherited paternally. This is the norm among certain coniferous plants, although not in pine trees and yews.[146] For Mytilids, paternal inheritance only occurs within males of the species.[147][148][149] It has been suggested that it occurs at a very low level in humans.[150] It was suggested in 2012, in an article in Current Biology, that mitochondria that shorten male lifespan stay in the system because they are inherited only through the mother. By contrast, natural selection weeds out mitochondria that reduce female survival as such mitochondria are less likely to be passed on to the next generation. Therefore, it is suggested that human females and female animals tend to live longer than males. The authors claim that this is a partial explanation.[151] Dr Tom Kirkwood, professor of ageing at Newcastle University, commented on the article "I certainly don't think this is a discovery that explains why women live five-to-six years longer than men."
Uniparental inheritance leads to little opportunity for genetic recombination between different lineages of mitochondria, although a single mitochondrion can contain 2–10 copies of its DNA.[125] For this reason, mitochondrial DNA is usually thought to reproduce by binary fission. What recombination does take place maintains genetic integrity rather than maintaining diversity. However, there are studies showing evidence of recombination in mitochondrial DNA. It is clear that the enzymes necessary for recombination are present in mammalian cells.[152] Further, evidence suggests that animal mitochondria can undergo recombination.[153] The data are a bit more controversial in humans, although indirect evidence of recombination exists.[154][155] If recombination does not occur, the whole mitochondrial DNA sequence represents a single haplotype, which makes it useful for studying the evolutionary history of populations.
Entities undergoing uniparental inheritance and with little to no recombination may be expected to be subject to Muller's ratchet, the inexorable accumulation of deleterious mutations until functionality is lost. Animal populations of mitochondria avoid this buildup through a developmental process known as the mtDNA bottleneck. The bottleneck exploits stochastic processes in the cell to increase in the cell-to-cell variability in mutant load as an organism develops: a single egg cell with some proportion of mutant mtDNA thus produces an embryo where different cells have different mutant loads. Cell-level selection may then act to remove those cells with more mutant mtDNA, leading to a stabilisation or reduction in mutant load between generations. The mechanism underlying the bottleneck is debated,[156][157][158] with a recent mathematical and experimental metastudy providing evidence for a combination of random partitioning of mtDNAs at cell divisions and random turnover of mtDNA molecules within the cell.[159]
DNA repair
Mitochondria can repair oxidative DNA damage by mechanisms that are analogous to those occurring in the cell nucleus. The proteins that are employed in mtDNA repair are encoded by nuclear genes, and are translocated to the mitochondria. The DNA repair pathways in mammalian mitochondria include base excision repair, double-strand break repair, direct reversal and mismatch repair.[160][161] Also DNA damages may be bypassed, rather than repaired, by translesion synthesis.
Of the several DNA repair process in mitochondria, the base excision repair pathway is the one that has been most comprehensively studied.[161] Base excision repair is carried out by a sequence of enzymatic catalyzed steps that include recognition and excision of a damaged DNA base, removal of the resulting abasic site, end processing, gap filling and ligation. A common damage in mtDNA that is repaired by base excision repair is 8-oxoguanine produced by the oxidation of guanine.[162]
Double-strand breaks can be repaired by homologous recombinational repair in both mammalian mtDNA[163] and plant mtDNA.[164] Double-strand breaks in mtDNA can also be repaired by microhomology-mediated end joining.[165] Although there is evidence for the repair processes of direct reversal and mismatch repair in mtDNA, these processes are still not well characterized.[161]
Lack of mitochondrial DNA
Eukaryotic cells typically have mitochondrial DNA; however, mitochondria that lack their own DNA have been found in a marine parasitic dinoflagellate from the genus Amoebophyra. This microorganism, A. cerati, has functional mitochondria that lack a genome.[166] In related species, the mitochondrial genome still has three genes, but in A. cerati only a single mitochondrial gene — the cytochrome c oxidase I gene (cox1) — is found, and it has migrated to the genome of the nucleus.[167]
Population genetic studies
The near-absence of genetic recombination in mitochondrial DNA makes it a useful source of information for scientists involved in population genetics and evolutionary biology.[168] Because all the mitochondrial DNA is inherited as a single unit, or haplotype, the relationships between mitochondrial DNA from different individuals can be represented as a gene tree. Patterns in these gene trees can be used to infer the evolutionary history of populations. The classic example of this is in human evolutionary genetics, where the molecular clock can be used to provide a recent date for mitochondrial Eve.[169][170] This is often interpreted as strong support for a recent modern human expansion out of Africa.[171] Another human example is the sequencing of mitochondrial DNA from Neanderthal bones. The relatively large evolutionary distance between the mitochondrial DNA sequences of Neanderthals and living humans has been interpreted as evidence for the lack of interbreeding between Neanderthals and anatomically modern humans.[172]
However, mitochondrial DNA reflects only the history of the females in a population and so may not represent the history of the population as a whole. This can be partially overcome by the use of paternal genetic sequences, such as the non-recombining region of the Y-chromosome.[171] In a broader sense, only studies that also include nuclear DNA can provide a comprehensive evolutionary history of a population.[173]
Recent measurements of the molecular clock for mitochondrial DNA[174] reported a value of 1 mutation every 7884 years dating back to the most recent common ancestor of humans and apes, which is consistent with estimates of mutation rates of autosomal DNA (10−8 per base per generation.[175]
Dysfunction and disease
Mitochondrial diseases
Damage and subsequent dysfunction in mitochondria is an important factor in a range of human diseases due to their influence in cell metabolism. Mitochondrial disorders often present themselves as neurological disorders, including autism.[16] They can also manifest as myopathy, diabetes, multiple endocrinopathy, and a variety of other systemic disorders.[176] Diseases caused by mutation in the mtDNA include Kearns–Sayre syndrome, MELAS syndrome and Leber's hereditary optic neuropathy.[177] In the vast majority of cases, these diseases are transmitted by a female to her children, as the zygote derives its mitochondria and hence its mtDNA from the ovum. Diseases such as Kearns-Sayre syndrome, Pearson syndrome, and progressive external ophthalmoplegia are thought to be due to large-scale mtDNA rearrangements, whereas other diseases such as MELAS syndrome, Leber's hereditary optic neuropathy, myoclonic epilepsy with ragged red fibers (MERRF), and others are due to point mutations in mtDNA.[176]
In other diseases, defects in nuclear genes lead to dysfunction of mitochondrial proteins. This is the case in Friedreich's ataxia, hereditary spastic paraplegia, and Wilson's disease.[178] These diseases are inherited in a dominance relationship, as applies to most other genetic diseases. A variety of disorders can be caused by nuclear mutations of oxidative phosphorylation enzymes, such as coenzyme Q10 deficiency and Barth syndrome.[176] Environmental influences may interact with hereditary predispositions and cause mitochondrial disease. For example, there may be a link between pesticide exposure and the later onset of Parkinson's disease.[179][180] Other pathologies with etiology involving mitochondrial dysfunction include schizophrenia, bipolar disorder, dementia, Alzheimer's disease,[181] [182]Parkinson's disease, epilepsy, stroke, cardiovascular disease, chronic fatigue syndrome, retinitis pigmentosa, and diabetes mellitus.[183][184]
Mitochondria-mediated oxidative stress plays a role in cardiomyopathy in Type 2 diabetics. Increased fatty acid delivery to the heart increases fatty acid uptake by cardiomyocytes, resulting in increased fatty acid oxidation in these cells. This process increases the reducing equivalents available to the electron transport chain of the mitochondria, ultimately increasing reactive oxygen species (ROS) production. ROS increases uncoupling proteins (UCPs) and potentiate proton leakage through the adenine nucleotide translocator (ANT), the combination of which uncouples the mitochondria. Uncoupling then increases oxygen consumption by the mitochondria, compounding the increase in fatty acid oxidation. This creates a vicious cycle of uncoupling; furthermore, even though oxygen consumption increases, ATP synthesis does not increase proportionally because the mitochondria are uncoupled. Less ATP availability ultimately results in an energy deficit presenting as reduced cardiac efficiency and contractile dysfunction. To compound the problem, impaired sarcoplasmic reticulum calcium release and reduced mitochondrial reuptake limits peak cytosolic levels of the important signaling ion during muscle contraction. Decreased intra-mitochondrial calcium concentration increases dehydrogenase activation and ATP synthesis. So in addition to lower ATP synthesis due to fatty acid oxidation, ATP synthesis is impaired by poor calcium signaling as well, causing cardiac problems for diabetics.[185]
Possible relationships to aging
Given the role of mitochondria as the cell's powerhouse, there may be some leakage of the high-energy electrons in the respiratory chain to form reactive oxygen species. This was thought to result in significant oxidative stress in the mitochondria with high mutation rates of mitochondrial DNA (mtDNA).[186] Hypothesized links between aging and oxidative stress are not new and were proposed in 1956,[187] which was later refined into the mitochondrial free radical theory of aging.[188] A vicious cycle was thought to occur, as oxidative stress leads to mitochondrial DNA mutations, which can lead to enzymatic abnormalities and further oxidative stress.
A number of changes can occur to mitochondria during the aging process.[189] Tissues from elderly patients show a decrease in enzymatic activity of the proteins of the respiratory chain.[190] However, mutated mtDNA can only be found in about 0.2% of very old cells.[191] Large deletions in the mitochondrial genome have been hypothesized to lead to high levels of oxidative stress and neuronal death in Parkinson's disease.[192]
In popular culture
Madeleine L'Engle's 1973 science fantasy novel A Wind in the Door prominently features the mitochondria of main character Charles Wallace Murry, as being inhabited by creatures known as the farandolae. The novel also features other characters traveling inside one of Murry's mitochondria.
The 1995 horror fiction novel Parasite Eve by Hideaki Sena depicts mitochondria as having some consciousness and mind control abilities, attempting to use these to overtake eukaryotes as the dominant life form. This text was adapted into an eponymous film, video game, and video game sequel all involving a similar premise.
In the Star Wars franchise, microorganisms referred to as "midi-chlorians" give some characters the ability to sense and use the Force. George Lucas, director of the 1999 film Star Wars: Episode I – The Phantom Menace, in which midi-chlorians were introduced, described them as "a loose depiction of mitochondria".[193] The non-fictional bacteria genus Midichloria was later named after the midi-chlorians of Star Wars.
As a result of the mitochondrion's prominence in modern American science education, the phrase "the mitochondria is the powerhouse of the cell" became an internet meme.
See also
References
- "Mitochondrion | Definition of Mitochondrion by Lexico". Lexico Dictionaries | English.
- Henze K, Martin W (November 2003). "Evolutionary biology: essence of mitochondria". Nature. 426 (6963): 127–128. Bibcode:2003Natur.426..127H. doi:10.1038/426127a. PMID 14614484.
- Karnkowska A, Vacek V, Zubáčová Z, Treitli SC, Petrželková R, Eme L, Novák L, Žárský V, Barlow LD, Herman EK, Soukal P, Hroudová M, Doležal P, Stairs CW, Roger AJ, Eliáš M, Dacks JB, Vlček Č, Hampl V (May 2016). "A Eukaryote without a Mitochondrial Organelle". Current Biology. 26 (10): 1274–1284. doi:10.1016/j.cub.2016.03.053. PMID 27185558.
- "Animal that doesn't need oxygen to survive discovered New Scientist". www.newscientist.com. Retrieved 2020-02-25.
- Yahalom, Dayana; Atkinson, Stephen D.; Neuhof, Moran; Chang, E. Sally; Philippe, Hervé; Cartwright, Paulyn; Bartholomew, Jerri L.; Huchon, Dorothée (2020-02-19). "A cnidarian parasite of salmon (Myxozoa: Henneguya) lacks a mitochondrial genome". Proceedings of the National Academy of Sciences. 117 (10): 5358–5363. doi:10.1073/pnas.1909907117. ISSN 0027-8424. PMC 7071853. PMID 32094163.
- "mitochondria". Online Etymology Dictionary.
- Campbell NA, Williamson B, Heyden RJ (2006). Biology: Exploring Life. Boston, Massachusetts: Pearson Prentice Hall. ISBN 978-0-13-250882-7.
- Siekevitz P (1957). "Powerhouse of the cell". Scientific American. 197 (1): 131–140. Bibcode:1957SciAm.197a.131S. doi:10.1038/scientificamerican0757-131.
- Wiemerslage L, Lee D (March 2016). "Quantification of mitochondrial morphology in neurites of dopaminergic neurons using multiple parameters". Journal of Neuroscience Methods. 262: 56–65. doi:10.1016/j.jneumeth.2016.01.008. PMC 4775301. PMID 26777473.
- McBride HM, Neuspiel M, Wasiak S (July 2006). "Mitochondria: more than just a powerhouse". Current Biology. 16 (14): R551–60. doi:10.1016/j.cub.2006.06.054. PMID 16860735.
- Valero T (2014). "Mitochondrial biogenesis: pharmacological approaches". Current Pharmaceutical Design. 20 (35): 5507–9. doi:10.2174/138161282035140911142118. hdl:10454/13341. PMID 24606795.
Mitochondrial biogenesis is therefore defined as the process via which cells increase their individual mitochondrial mass [3]. ... Mitochondrial biogenesis occurs by growth and division of pre-existing organelles and is temporally coordinated with cell cycle events [1].
- Sanchis-Gomar F, García-Giménez JL, Gómez-Cabrera MC, Pallardó FV (2014). "Mitochondrial biogenesis in health and disease. Molecular and therapeutic approaches". Current Pharmaceutical Design. 20 (35): 5619–33. doi:10.2174/1381612820666140306095106. PMID 24606801.
Mitochondrial biogenesis (MB) is the essential mechanism by which cells control the number of mitochondria
- Gardner A, Boles RG (2005). "Is a 'Mitochondrial Psychiatry' in the Future? A Review". Curr. Psychiatry Rev. 1 (3): 255–271. doi:10.2174/157340005774575064.
- Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL (June 2001). "Mitochondrial dysfunction in cardiac disease: ischemia--reperfusion, aging, and heart failure". Journal of Molecular and Cellular Cardiology. 33 (6): 1065–89. doi:10.1006/jmcc.2001.1378. PMID 11444914.
- Dorn GW, Vega RB, Kelly DP (October 2015). "Mitochondrial biogenesis and dynamics in the developing and diseased heart". Genes & Development. 29 (19): 1981–91. doi:10.1101/gad.269894.115. PMC 4604339. PMID 26443844.
- Griffiths KK, Levy RJ (2017). "Evidence of Mitochondrial Dysfunction in Autism: Biochemical Links, Genetic-Based Associations, and Non-Energy-Related Mechanisms". Oxidative Medicine and Cellular Longevity. 2017: 4314025. doi:10.1155/2017/4314025. PMC 5467355. PMID 28630658.
- Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P (2005). Molecular Biology of the Cell. New York: Garland Publishing Inc. ISBN 978-0-8153-4105-5.
- Voet D, Voet JG, Pratt CW (2006). Fundamentals of Biochemistry (2nd ed.). John Wiley and Sons, Inc. pp. 547, 556. ISBN 978-0-471-21495-3.
- Andersson SG, Karlberg O, Canbäck B, Kurland CG (January 2003). "On the origin of mitochondria: a genomics perspective". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 358 (1429): 165–77, discussion 177–9. doi:10.1098/rstb.2002.1193. PMC 1693097. PMID 12594925.
- Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, Wiley S, Murphy AN, Gaucher SP, Capaldi RA, Gibson BW, Ghosh SS (March 2003). "Characterization of the human heart mitochondrial proteome". Nature Biotechnology. 21 (3): 281–6. doi:10.1038/nbt793. PMID 12592411.
- Zhang J, Li X, Mueller M, Wang Y, Zong C, Deng N, Vondriska TM, Liem DA, Yang JI, Korge P, Honda H, Weiss JN, Apweiler R, Ping P (April 2008). "Systematic characterization of the murine mitochondrial proteome using functionally validated cardiac mitochondria". Proteomics. 8 (8): 1564–75. doi:10.1002/pmic.200700851. PMC 2799225. PMID 18348319.
- Zhang J, Liem DA, Mueller M, Wang Y, Zong C, Deng N, Vondriska TM, Korge P, Drews O, Maclellan WR, Honda H, Weiss JN, Apweiler R, Ping P (June 2008). "Altered proteome biology of cardiac mitochondria under stress conditions". Journal of Proteome Research. 7 (6): 2204–14. doi:10.1021/pr070371f. PMC 3805274. PMID 18484766.
- Ernster L, Schatz G (December 1981). "Mitochondria: a historical review". The Journal of Cell Biology. 91 (3 Pt 2): 227s–255s. doi:10.1083/jcb.91.3.227s. PMC 2112799. PMID 7033239.
- Altmann, R. 1890 . Die Elementarorganismen und ihre Beziehungen zu den Zellen. Veit, Leipzig, .
- Benda C (1898). "Ueber die Spermatogenese der Vertebraten und höherer Evertebraten. II. Theil: Die Histiogenese der Spermien". Arch. Anal. Physiol.: 393–398.
- Ernster's citation Meves, Friedrich (May 1908). "Die Chondriosomen als Träger erblicher Anlagen. Cytologische Studien am Hühnerembryo". Archiv für Mikroskopische Anatomie. 72 (1): 816–867. doi:10.1007/BF02982402. is wrong, correct citation is Meves, Friedrich (1904). "Über das Vorkommen von Mitochondrien bezw. Chondromiten in Pflanzenzellen". Ber. Dtsch. Bot. Ges. 22: 284–286., cited in Meves' 1908 paper and in Schmidt, Ernst Willy (1913). "Pflanzliche Mitochondrien". Progressus Rei Botanicae. 4: 164–183. Retrieved 21 September 2012., with confirmation of Nymphaea alba
- Martin WF, Garg S, Zimorski V (September 2015). "Endosymbiotic theories for eukaryote origin". Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences. 370 (1678): 20140330. doi:10.1098/rstb.2014.0330. PMC 4571569. PMID 26323761.
- Margulis L, Sagan D (1986). Origins of Sex. Three Billion Years of Genetic Recombination. New Haven: Yale University Press. pp. 69–71, 87. ISBN 978-0-300-03340-3.
- Martin WF, Müller M (2007). Origin of mitochondria and hydrogenosomes. Heidelberg: Springer Verlag.
- Emelyanov VV (April 2003). "Mitochondrial connection to the origin of the eukaryotic cell". European Journal of Biochemistry. 270 (8): 1599–1618. doi:10.1046/j.1432-1033.2003.03499.x. PMID 12694174.
- Müller M, Martin W (May 1999). "The genome of Rickettsia prowazekii and some thoughts on the origin of mitochondria and hydrogenosomes" (PDF). BioEssays. 21 (5): 377–381. doi:10.1002/(sici)1521-1878(199905)21:5<377::aid-bies4>3.0.co;2-w. PMID 10376009.
- Gray MW, Burger G, Lang BF (March 1999). "Mitochondrial evolution". Science. 283 (5407): 1476–1481. Bibcode:1999Sci...283.1476G. doi:10.1126/science.283.5407.1476. PMID 10066161.
- Thrash JC, Boyd A, Huggett MJ, Grote J, Carini P, Yoder RJ, Robbertse B, Spatafora JW, Rappé MS, Giovannoni SJ (2011-06-14). "Phylogenomic evidence for a common ancestor of mitochondria and the SAR11 clade". Scientific Reports. 1 (1): 13. Bibcode:2011NatSR...1E..13T. doi:10.1038/srep00013. PMC 3216501. PMID 22355532.
- Martijn J, Vosseberg J, Guy L, Offre P, Ettema TJ (April 2018). "Deep mitochondrial origin outside the sampled alphaproteobacteria". Nature. 557 (7703): 101–105. Bibcode:2018Natur.557..101M. doi:10.1038/s41586-018-0059-5. PMID 29695865.
- Fan, Lu; Wu, Dingfeng; Goremykin, Vadim; Xiao, Jing; Xu, Yanbing; Garg, Sriram; Zhang, Chuanlun; Martin, William F.; Zhu, Ruixin (2019-07-26). "Mitochondria branch within Alphaproteobacteria". bioRxiv: 715870. doi:10.1101/715870.
- Ferla MP, Thrash JC, Giovannoni SJ, Patrick WM (2013). "New rRNA gene-based phylogenies of the Alphaproteobacteria provide perspective on major groups, mitochondrial ancestry and phylogenetic instability". PLOS One. 8 (12): e83383. doi:10.1371/journal.pone.0083383. PMC 3859672. PMID 24349502.
- O'Brien TW (September 2003). "Properties of human mitochondrial ribosomes". IUBMB Life. 55 (9): 505–513. doi:10.1080/15216540310001626610. PMID 14658756.
- Sagan L (March 1967). "On the origin of mitosing cells". Journal of Theoretical Biology. 14 (3): 255–274. doi:10.1016/0022-5193(67)90079-3. PMID 11541392.
- Emelyanov VV (February 2001). "Rickettsiaceae, rickettsia-like endosymbionts, and the origin of mitochondria". Bioscience Reports. 21 (1): 1–17. doi:10.1023/A:1010409415723. PMID 11508688.
- Feng DF, Cho G, Doolittle RF (November 1997). "Determining divergence times with a protein clock: update and reevaluation". Proceedings of the National Academy of Sciences of the United States of America. 94 (24): 13028–13033. Bibcode:1997PNAS...9413028F. doi:10.1073/pnas.94.24.13028. PMC 24257. PMID 9371794.
- Cavalier-Smith T (1991). "Archamoebae: the ancestral eukaryotes?". Bio Systems. 25 (1–2): 25–38. doi:10.1016/0303-2647(91)90010-I. PMID 1854912.
- Danovaro R (2010). "The first metazoa living in permanently anoxic conditions". BMC Biology. 8: 30. doi:10.1186/1741-7007-8-30. PMC 2907586. PMID 20370908.
- Andy Coghaln: Zoologger: The mud creature that lives without oxygen, NewScientist, April 7, 2010
- Yahalomi D, Atkinson SD, Neuhof M, Chang ES, Philippe H, Cartwright P, Bartholomew JL, Huchon D (2020). "A cnidarian parasite of salmon (Myxozoa: Henneguya) lacks a mitochondrial genome". Proc Natl Acad Sci U S A. 117 (10): 5358–5363. doi:10.1073/pnas.1909907117. PMC 7071853. PMID 32094163.
- Shiflett, AM; Johnson, PJ (2010). "Mitochondrion-related organelles in eukaryotic protists". Annual Review of Microbiology. 64: 409–29. doi:10.1146/annurev.micro.62.081307.162826. PMC 3208401. PMID 20528687.
- Karnkowska A, Vacek V, Zubáčová Z, Treitli SC, Petrželková R, Eme L, Novák L, Žárský V, Barlow LD, Herman EK, Soukal P, Hroudová M, Doležal P, Stairs CW, Roger AJ, Eliáš M, Dacks JB, Vlček Č, Hampl V (May 2016). "A Eukaryote without a Mitochondrial Organelle". Current Biology. 26 (10): 1274–1284. doi:10.1016/j.cub.2016.03.053. PMID 27185558.
- "Mitochondrion – much more than an energy converter". British Society for Cell Biology. Retrieved 19 August 2013.
- Blachly-Dyson E, Forte M (September 2001). "VDAC channels". IUBMB Life. 52 (3–5): 113–8. doi:10.1080/15216540152845902. PMID 11798022.
- Hoogenboom BW, Suda K, Engel A, Fotiadis D (July 2007). "The supramolecular assemblies of voltage-dependent anion channels in the native membrane". Journal of Molecular Biology. 370 (2): 246–55. doi:10.1016/j.jmb.2007.04.073. PMID 17524423.
- Zeth K (June 2010). "Structure and evolution of mitochondrial outer membrane proteins of beta-barrel topology". Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1797 (6–7): 1292–9. doi:10.1016/j.bbabio.2010.04.019. PMID 20450883.
- Herrmann JM, Neupert W (April 2000). "Protein transport into mitochondria". Current Opinion in Microbiology. 3 (2): 210–4. doi:10.1016/S1369-5274(00)00077-1. PMID 10744987.
- Chipuk JE, Bouchier-Hayes L, Green DR (August 2006). "Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario". Cell Death and Differentiation. 13 (8): 1396–1402. doi:10.1038/sj.cdd.4401963. PMID 16710362.
- Hayashi T, Rizzuto R, Hajnoczky G, Su TP (February 2009). "MAM: more than just a housekeeper". Trends in Cell Biology. 19 (2): 81–88. doi:10.1016/j.tcb.2008.12.002. PMC 2750097. PMID 19144519.
- Schenkel LC, Bakovic M (January 2014). "Formation and Regulation of Mitochondrial Membranes". International Journal of Cell Biology. 2014: 709828. doi:10.1155/2014/709828. PMC 3918842. PMID 24578708.
- McMillin JB, Dowhan W (December 2002). "Cardiolipin and apoptosis". Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids. 1585 (2–3): 97–107. doi:10.1016/S1388-1981(02)00329-3. PMID 12531542.
- Youle RJ, van der Bliek AM (2012). "Mitochondrial Fission, Fusion, and Stress". Science. 337 (6098): 1062–1065. Bibcode:2012Sci...337.1062Y. doi:10.1126/science.1219855. PMC 4762028. PMID 22936770.
- Cserép C, Pósfai B, Schwarcz AD, Dénes Á (2018). "Mitochondrial Ultrastructure Is Coupled to Synaptic Performance at Axonal Release Sites". eNeuro. 5 (1). doi:10.1523/ENEURO.0390-17.2018. PMC 5788698. PMID 29383328.
- Mannella CA (2006). "Structure and dynamics of the mitochondrial inner membrane cristae". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1763 (5–6): 542–548. doi:10.1016/j.bbamcr.2006.04.006. PMID 16730811.
- Thar R, Kühl M (September 2004). "Propagation of electromagnetic radiation in mitochondria?" (PDF). Journal of Theoretical Biology. 230 (2): 261–270. doi:10.1016/j.jtbi.2004.05.021. PMID 15302557.
- Bogenhagen DF (September 2012). "Mitochondrial DNA nucleoid structure". Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms. 1819 (9–10): 914–20. doi:10.1016/j.bbagrm.2011.11.005. PMID 22142616.
- Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (April 1981). "Sequence and organization of the human mitochondrial genome". Nature. 290 (5806): 457–465. Bibcode:1981Natur.290..457A. doi:10.1038/290457a0. PMID 7219534.
- Rizzuto R, Marchi S, Bonora M, Aguiari P, Bononi A, De Stefani D, Giorgi C, Leo S, Rimessi A, Siviero R, Zecchini E, Pinton P (November 2009). "Ca(2+) transfer from the ER to mitochondria: when, how and why". Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1787 (11): 1342–1351. doi:10.1016/j.bbabio.2009.03.015. PMC 2730423. PMID 19341702.
- de Brito OM, Scorrano L (August 2010). "An intimate liaison: spatial organization of the endoplasmic reticulum-mitochondria relationship". The EMBO Journal. 29 (16): 2715–2723. doi:10.1038/emboj.2010.177. PMC 2924651. PMID 20717141.
- Vance JE, Shiao YJ (1996). "Intracellular trafficking of phospholipids: import of phosphatidylserine into mitochondria". Anticancer Research. 16 (3B): 1333–1339. PMID 8694499.
- Lebiedzinska M, Szabadkai G, Jones AW, Duszynski J, Wieckowski MR (October 2009). "Interactions between the endoplasmic reticulum, mitochondria, plasma membrane and other subcellular organelles". The International Journal of Biochemistry & Cell Biology. 41 (10): 1805–1816. doi:10.1016/j.biocel.2009.02.017. PMID 19703651.
- Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS (January 2008). "Fission and selective fusion govern mitochondrial segregation and elimination by autophagy". The EMBO Journal. 27 (2): 433–446. doi:10.1038/sj.emboj.7601963. PMC 2234339. PMID 18200046.
- Osman C, Voelker DR, Langer T (January 2011). "Making heads or tails of phospholipids in mitochondria". The Journal of Cell Biology. 192 (1): 7–16. doi:10.1083/jcb.201006159. PMC 3019561. PMID 21220505.
- Kornmann B, Currie E, Collins SR, Schuldiner M, Nunnari J, Weissman JS, Walter P (July 2009). "An ER-mitochondria tethering complex revealed by a synthetic biology screen". Science. 325 (5939): 477–481. Bibcode:2009Sci...325..477K. doi:10.1126/science.1175088. PMC 2933203. PMID 19556461.
- Rusiñol AE, Cui Z, Chen MH, Vance JE (November 1994). "A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins". The Journal of Biological Chemistry. 269 (44): 27494–27502. PMID 7961664.
- Santulli G, Marks AR (2015). "Essential Roles of Intracellular Calcium Release Channels in Muscle, Brain, Metabolism, and Aging". Current Molecular Pharmacology. 8 (2): 206–222. doi:10.2174/1874467208666150507105105. PMID 25966694.
- Kopach O, Kruglikov I, Pivneva T, Voitenko N, Fedirko N (May 2008). "Functional coupling between ryanodine receptors, mitochondria and Ca(2+) ATPases in rat submandibular acinar cells". Cell Calcium. 43 (5): 469–481. doi:10.1016/j.ceca.2007.08.001. PMID 17889347.
- Csordás G, Hajnóczky G (April 2001). "Sorting of calcium signals at the junctions of endoplasmic reticulum and mitochondria". Cell Calcium. 29 (4): 249–262. doi:10.1054/ceca.2000.0191. PMID 11243933.
- Decuypere JP, Monaco G, Bultynck G, Missiaen L, De Smedt H, Parys JB (May 2011). "The IP(3) receptor-mitochondria connection in apoptosis and autophagy". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1813 (5): 1003–1013. doi:10.1016/j.bbamcr.2010.11.023. PMID 21146562.
- Hajnóczky G, Csordás G, Yi M (2011). "Old players in a new role: mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria". Cell Calcium. 32 (5–6): 363–377. doi:10.1016/S0143416002001872. PMID 12543096.
- Marriott KS, Prasad M, Thapliyal V, Bose HS (December 2012). "σ-1 receptor at the mitochondrial-associated endoplasmic reticulum membrane is responsible for mitochondrial metabolic regulation". The Journal of Pharmacology and Experimental Therapeutics. 343 (3): 578–586. doi:10.1124/jpet.112.198168. PMC 3500540. PMID 22923735.
- Hayashi T, Su TP (November 2007). "Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival". Cell. 131 (3): 596–610. doi:10.1016/j.cell.2007.08.036. PMID 17981125.
- Csordás et al., Trends Cell Biol. 2018 Jul;28(7):523-540. doi: 10.1016/j.tcb.2018.02.009. Epub 2018 Mar 24.
- Cserép C, Pósfai B, Lénárt N, Fekete R, László ZI, Lele Z, et al. (January 2020). "Microglia monitor and protect neuronal function through specialized somatic purinergic junctions". Science. 367 (6477): 528–537. doi:10.1126/science.aax6752. PMID 31831638.
- "Animal that doesn't need oxygen to survive discovered New Scientist". www.newscientist.com. Retrieved 2020-02-25.
- The eukaryote Giardia lamblia, for example, does not contain mitochondria, but does have a mitochondrial-like gene, suggesting that it once included either mitochondria or an endosymbiotic progenitor of it Roger AJ, Svärd SG, Tovar J, Clark CG, Smith MW, Gillin FD, Sogin ML (January 1998). "A mitochondrial-like chaperonin 60 gene in Giardia lamblia: evidence that diplomonads once harbored an endosymbiont related to the progenitor of mitochondria". Proceedings of the National Academy of Sciences of the United States of America. 95 (1): 229–234. Bibcode:1998PNAS...95..229R. doi:10.1073/pnas.95.1.229. PMC 18184. PMID 9419358.
- Logan DC (June 2010). "Mitochondrial fusion, division and positioning in plants". Biochemical Society Transactions. 38 (3): 789–95. doi:10.1042/bst0380789. PMID 20491666.
- das Neves RP, Jones NS, Andreu L, Gupta R, Enver T, Iborra FJ (December 2010). Weissman JS (ed.). "Connecting variability in global transcription rate to mitochondrial variability". PLOS Biology. 8 (12): e1000560. doi:10.1371/journal.pbio.1000560. PMC 3001896. PMID 21179497.
- Johnston IG, Gaal B, Neves RP, Enver T, Iborra FJ, Jones NS (2012). Haugh JM (ed.). "Mitochondrial variability as a source of extrinsic cellular noise". PLOS Computational Biology. 8 (3): e1002416. arXiv:1107.4499. Bibcode:2012PLSCB...8E2416J. doi:10.1371/journal.pcbi.1002416. PMC 3297557. PMID 22412363.
- Rappaport L, Oliviero P, Samuel JL (1998). "Cytoskeleton and mitochondrial morphology and function". Mol. Cell. Biochem. 184: 101–105. doi:10.1023/A:1006843113166.
- Hoitzing H, Johnston IG, Jones NS (June 2015). "What is the function of mitochondrial networks? A theoretical assessment of hypotheses and proposal for future research". BioEssays. 37 (6): 687–700. doi:10.1002/bies.201400188. PMC 4672710. PMID 25847815.
- Soltys BJ, Gupta RS (1992). "Interrelationships of endoplasmic reticulum, mitochondria, intermediate filaments, and microtubules--a quadruple fluorescence labeling study". Biochemistry and Cell Biology. 70 (10–11): 1174–1186. doi:10.1139/o92-163. PMID 1363623.
- Tang HL, Lung HL, Wu KC, Le AH, Tang HM, Fung MC (February 2008). "Vimentin supports mitochondrial morphology and organization". The Biochemical Journal. 410 (1): 141–146. doi:10.1042/BJ20071072. PMID 17983357.
- Schmidt-Rohr K (2020). "Oxygen Is the High-Energy Molecule Powering Complex Multicellular Life: Fundamental Corrections to Traditional Bioenergetics". ACS Omega. 5 (5): 2221–2233. doi:10.1021/acsomega.9b03352. PMC 7016920. PMID 32064383.
- Rich PR (December 2003). "The molecular machinery of Keilin's respiratory chain". Biochemical Society Transactions. 31 (Pt 6): 1095–1105. doi:10.1042/BST0311095. PMID 14641005.
- Stoimenova M, Igamberdiev AU, Gupta KJ, Hill RD (July 2007). "Nitrite-driven anaerobic ATP synthesis in barley and rice root mitochondria". Planta. 226 (2): 465–474. doi:10.1007/s00425-007-0496-0. PMID 17333252.
- Stryer L (1995). "Citric acid cycle.". In: Biochemistry (Fourth ed.). New York: W.H. Freeman and Company. pp. 509–527, 569–579, 614–616, 638–641, 732–735, 739–748, 770–773. ISBN 0-7167-2009-4.
- King A, Selak MA, Gottlieb E (August 2006). "Succinate dehydrogenase and fumarate hydratase: linking mitochondrial dysfunction and cancer". Oncogene. 25 (34): 4675–4682. doi:10.1038/sj.onc.1209594. PMID 16892081.
- Huang H, Manton KG (May 2004). "The role of oxidative damage in mitochondria during aging: a review" (PDF). Frontiers in Bioscience. 9 (1–3): 1100–1117. doi:10.2741/1298. PMID 14977532.
- Mitchell P, Moyle J (January 1967). "Chemiosmotic hypothesis of oxidative phosphorylation". Nature. 213 (5072): 137–139. Bibcode:1967Natur.213..137M. doi:10.1038/213137a0. PMID 4291593.
- Mitchell P (June 1967). "Proton current flow in mitochondrial systems". Nature. 214 (5095): 1327–1328. Bibcode:1967Natur.214.1327M. doi:10.1038/2141327a0. PMID 6056845.
- Nobel Foundation. "Chemistry 1997". Retrieved 2007-12-16.
- Mozo J, Emre Y, Bouillaud F, Ricquier D, Criscuolo F (November 2005). "Thermoregulation: what role for UCPs in mammals and birds?". Bioscience Reports. 25 (3–4): 227–249. doi:10.1007/s10540-005-2887-4. PMID 16283555.
- Nicholls DG, Lindberg O (September 1973). "Brown-adipose-tissue mitochondria. The influence of albumin and nucleotides on passive ion permeabilities". European Journal of Biochemistry. 37 (3): 523–530. doi:10.1111/j.1432-1033.1973.tb03014.x. PMID 4777251.
- Santulli G, Xie W, Reiken SR, Marks AR (September 2015). "Mitochondrial calcium overload is a key determinant in heart failure". Proceedings of the National Academy of Sciences of the United States of America. 112 (36): 11389–11394. Bibcode:2015PNAS..11211389S. doi:10.1073/pnas.1513047112. PMC 4568687. PMID 26217001.
- Siegel GJ, Agranoff BW, Fisher SK, Albers RW, Uhler MD, eds. (1999). Basic Neurochemistry (6 ed.). Lippincott Williams & Wilkins. ISBN 978-0-397-51820-3. Illustrations by Lorie M. Gavulic
- Rossier MF (August 2006). "T channels and steroid biosynthesis: in search of a link with mitochondria". Cell Calcium. 40 (2): 155–164. doi:10.1016/j.ceca.2006.04.020. PMID 16759697.
- Brighton CT, Hunt RM (May 1974). "Mitochondrial calcium and its role in calcification. Histochemical localization of calcium in electron micrographs of the epiphyseal growth plate with K-pyroantimonate". Clinical Orthopaedics and Related Research. 100 (5): 406–416. doi:10.1097/00003086-197405000-00057. PMID 4134194.
- Brighton CT, Hunt RM (July 1978). "The role of mitochondria in growth plate calcification as demonstrated in a rachitic model". The Journal of Bone and Joint Surgery. American Volume. 60 (5): 630–639. doi:10.2106/00004623-197860050-00007. PMID 681381.
- Pizzo P, Pozzan T (October 2007). "Mitochondria-endoplasmic reticulum choreography: structure and signaling dynamics". Trends in Cell Biology. 17 (10): 511–517. doi:10.1016/j.tcb.2007.07.011. PMID 17851078.
- Miller RJ (March 1, 1998). "Mitochondria – the kraken wakes!". Trends Neurosci. 21 (3): 95–97. doi:10.1016/S0166-2236(97)01206-X. PMID 9530913.
- Santulli G, Pagano G, Sardu C, Xie W, Reiken S, D'Ascia SL, Cannone M, Marziliano N, Trimarco B, Guise TA, Lacampagne A, Marks AR (May 2015). "Calcium release channel RyR2 regulates insulin release and glucose homeostasis". The Journal of Clinical Investigation. 125 (5): 1968–1978. doi:10.1172/JCI79273. PMC 4463204. PMID 25844899.
- Schwarzländer M, Logan DC, Johnston IG, Jones NS, Meyer AJ, Fricker MD, Sweetlove LJ (March 2012). "Pulsing of membrane potential in individual mitochondria: a stress-induced mechanism to regulate respiratory bioenergetics in Arabidopsis". The Plant Cell. 24 (3): 1188–1201. doi:10.1105/tpc.112.096438. PMC 3336130. PMID 22395486.
- Ivannikov MV, Macleod GT (June 2013). "Mitochondrial free Ca²⁺ levels and their effects on energy metabolism in Drosophila motor nerve terminals". Biophysical Journal. 104 (11): 2353–2361. Bibcode:2013BpJ...104.2353I. doi:10.1016/j.bpj.2013.03.064. PMC 3672877. PMID 23746507.
- Li X, Fang P, Mai J, Choi ET, Wang H, Yang XF (February 2013). "Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers". Journal of Hematology & Oncology. 6 (19): 19. doi:10.1186/1756-8722-6-19. PMC 3599349. PMID 23442817.
- Green DR (September 1998). "Apoptotic pathways: the roads to ruin". Cell. 94 (6): 695–698. doi:10.1016/S0092-8674(00)81728-6. PMID 9753316.
- Hajnóczky G, Csordás G, Das S, Garcia-Perez C, Saotome M, Sinha Roy S, Yi M (2006). "Mitochondrial calcium signalling and cell death: approaches for assessing the role of mitochondrial Ca2+ uptake in apoptosis". Cell Calcium. 40 (5–6): 553–560. doi:10.1016/j.ceca.2006.08.016. PMC 2692319. PMID 17074387.
- Oh-hama T (August 1997). "Evolutionary consideration on 5-aminolevulinate synthase in nature". Origins of Life and Evolution of the Biosphere. 27 (4): 405–412. doi:10.1023/A:1006583601341. PMID 9249985.
- Klinge CM (December 2008). "Estrogenic control of mitochondrial function and biogenesis". Journal of Cellular Biochemistry. 105 (6): 1342–1351. doi:10.1002/jcb.21936. PMC 2593138. PMID 18846505.
- Alvarez-Delgado C, Mendoza-Rodríguez CA, Picazo O, Cerbón M (August 2010). "Different expression of alpha and beta mitochondrial estrogen receptors in the aging rat brain: interaction with respiratory complex V". Experimental Gerontology. 45 (7–8): 580–585. doi:10.1016/j.exger.2010.01.015. PMID 20096765.
- Pavón N, Martínez-Abundis E, Hernández L, Gallardo-Pérez JC, Alvarez-Delgado C, Cerbón M, Pérez-Torres I, Aranda A, Chávez E (October 2012). "Sexual hormones: effects on cardiac and mitochondrial activity after ischemia-reperfusion in adult rats. Gender difference". The Journal of Steroid Biochemistry and Molecular Biology. 132 (1–2): 135–146. doi:10.1016/j.jsbmb.2012.05.003. PMID 22609314.
- Naffah et. al, https://doi.org/10.1016/j.redox.2019.101255
- Weinberg F, Chandel NS (October 2009). "Mitochondrial metabolism and cancer". Annals of the New York Academy of Sciences. 1177 (1): 66–73. Bibcode:2009NYASA1177...66W. doi:10.1111/j.1749-6632.2009.05039.x. PMID 19845608.
- Moreno-Sánchez R, Rodríguez-Enríquez S, Marín-Hernández A, Saavedra E (March 2007). "Energy metabolism in tumor cells". The FEBS Journal. 274 (6): 1393–1418. doi:10.1111/j.1742-4658.2007.05686.x. PMID 17302740.
- Mistry JJ, Marlein CR, Moore J, Hellmich C, Wojtowicz EE, Smith JG, Macaulay I, Sun Y, Morfakis A, Patterson A, Horton RH, Divekar D, Morris CJ, Haestier A, Di Palma F, Beraza N, Bowles KM, Rushworth SA (November 2019). "ROS-mediated PI3K activation drives mitochondrial transfer from stromal cells to hematopoietic stem cells in response to infection". PNAS. 116 (49): 24610–24619. doi:10.1073/pnas.1913278116. PMC 6900710. PMID 31727843.
- Pedersen PL (December 1994). "ATP synthase. The machine that makes ATP". Current Biology. 4 (12): 1138–1141. doi:10.1016/S0960-9822(00)00257-8. PMID 7704582.
- Pattappa G, Heywood HK, de Bruijn JD, Lee DA (October 2011). "The metabolism of human mesenchymal stem cells during proliferation and differentiation". Journal of Cellular Physiology. 226 (10): 2562–2570. doi:10.1002/jcp.22605. PMID 21792913.
- Agarwal B (June 2011). "A role for anions in ATP synthesis and its molecular mechanistic interpretation". Journal of Bioenergetics and Biomembranes. 43 (3): 299–310. doi:10.1007/s10863-011-9358-3. PMID 21647635.
- Sweet S, Singh G (July 1999). "Changes in mitochondrial mass, membrane potential, and cellular adenosine triphosphate content during the cell cycle of human leukemic (HL-60) cells". Journal of Cellular Physiology. 180 (1): 91–96. doi:10.1002/(SICI)1097-4652(199907)180:1<91::AID-JCP10>3.0.CO;2-6. PMID 10362021.
- Chan DC (June 2006). "Mitochondria: dynamic organelles in disease, aging, and development". Cell. 125 (7): 1241–1252. doi:10.1016/j.cell.2006.06.010. PMID 16814712.
- Wiesner RJ, Rüegg JC, Morano I (March 1992). "Counting target molecules by exponential polymerase chain reaction: copy number of mitochondrial DNA in rat tissues". Biochemical and Biophysical Research Communications. 183 (2): 553–559. doi:10.1016/0006-291X(92)90517-O. PMID 1550563.
- Fukuhara H, Sor F, Drissi R, Dinouël N, Miyakawa I, Rousset S, Viola AM (April 1993). "Linear mitochondrial DNAs of yeasts: frequency of occurrence and general features". Molecular and Cellular Biology. 13 (4): 2309–2314. doi:10.1128/mcb.13.4.2309. PMC 359551. PMID 8455612.
- Bernardi G (December 1978). "Intervening sequences in the mitochondrial genome". Nature. 276 (5688): 558–559. Bibcode:1978Natur.276..558B. doi:10.1038/276558a0. PMID 214710.
- Hebbar SK, Belcher SM, Perlman PS (April 1992). "A maturase-encoding group IIA intron of yeast mitochondria self-splices in vitro". Nucleic Acids Research. 20 (7): 1747–1754. doi:10.1093/nar/20.7.1747. PMC 312266. PMID 1579468.
- Gray MW, Lang BF, Cedergren R, Golding GB, Lemieux C, Sankoff D, Turmel M, Brossard N, Delage E, Littlejohn TG, Plante I, Rioux P, Saint-Louis D, Zhu Y, Burger G (February 1998). "Genome structure and gene content in protist mitochondrial DNAs". Nucleic Acids Research. 26 (4): 865–878. doi:10.1093/nar/26.4.865. PMC 147373. PMID 9461442.
- Gray MW, Lang BF, Burger G (2004). "Mitochondria of protists". Annual Review of Genetics. 38: 477–524. doi:10.1146/annurev.genet.37.110801.142526. PMID 15568984.
- Telonis AG, Loher P, Kirino Y, Rigoutsos I (2014). "Nuclear and mitochondrial tRNA-lookalikes in the human genome". Frontiers in Genetics. 5: 344. doi:10.3389/fgene.2014.00344. PMC 4189335. PMID 25339973.
- Shao R, Kirkness EF, Barker SC (May 2009). "The single mitochondrial chromosome typical of animals has evolved into 18 minichromosomes in the human body louse, Pediculus humanus". Genome Research. 19 (5): 904–912. doi:10.1101/gr.083188.108. PMC 2675979. PMID 19336451.
- Crick FH, Orgel LE (1973). "Directed panspermia" (PDF). Icarus. 19 (3): 341–346. Bibcode:1973Icar...19..341C. doi:10.1016/0019-1035(73)90110-3.
p. 344: It is a little surprising that organisms with somewhat different codes do not coexist.
Further discussion. - Barrell BG, Bankier AT, Drouin J (November 1979). "A different genetic code in human mitochondria". Nature. 282 (5735): 189–194. Bibcode:1979Natur.282..189B. doi:10.1038/282189a0. PMID 226894.
- Mitochondrial Genetic Code in Taxonomy Tree. NCBI
- Elzanowski, Andrzej and Ostell, Jim. The Genetic Codes. NCBI
- Jukes TH, Osawa S (December 1990). "The genetic code in mitochondria and chloroplasts". Experientia. 46 (11–12): 1117–1126. doi:10.1007/BF01936921. PMID 2253709.
- Hiesel R, Wissinger B, Schuster W, Brennicke A (December 1989). "RNA editing in plant mitochondria". Science. 246 (4937): 1632–1634. Bibcode:1989Sci...246.1632H. doi:10.1126/science.2480644. PMID 2480644.
- Abascal F, Posada D, Knight RD, Zardoya R (May 2006). "Parallel evolution of the genetic code in arthropod mitochondrial genomes". PLOS Biology. 4 (5): e127. doi:10.1371/journal.pbio.0040127. PMC 1440934. PMID 16620150.
- Henriquez FL, Richards TA, Roberts F, McLeod R, Roberts CW (February 2005). "The unusual mitochondrial compartment of Cryptosporidium parvum". Trends in Parasitology. 21 (2): 68–74. doi:10.1016/j.pt.2004.11.010. PMID 15664529.
- Pfeiffer RF (2012). Parkinson's Disease. CRC Press. p. 583. ISBN 9781439807149.
- Seo AY, Joseph AM, Dutta D, Hwang JC, Aris JP, Leeuwenburgh C (August 2010). "New insights into the role of mitochondria in aging: mitochondrial dynamics and more". Journal of Cell Science. 123 (Pt 15): 2533–2542. doi:10.1242/jcs.070490. PMC 2912461. PMID 20940129.
- Hu GB (August 2014). "Whole cell cryo-electron tomography suggests mitochondria divide by budding". Microscopy and Microanalysis. 20 (4): 1180–1187. Bibcode:2014MiMic..20.1180H. doi:10.1017/S1431927614001317. PMID 24870811.
- Kimball, J.W. (2006) "Sexual Reproduction in Humans: Copulation and Fertilization," Kimball's Biology Pages (based on Biology, 6th ed., 1996)
- Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G (November 1999). "Ubiquitin tag for sperm mitochondria". Nature. 402 (6760): 371–372. Bibcode:1999Natur.402..371S. doi:10.1038/46466. PMID 10586873. Discussed in Science News.
- Mogensen HL (1996). "The Hows and Whys of Cytoplasmic Inheritance in Seed Plants". American Journal of Botany. 83 (3): 383–404. doi:10.2307/2446172. JSTOR 2446172.
- Zouros E (December 2000). "The exceptional mitochondrial DNA system of the mussel family Mytilidae". Genes & Genetic Systems. 75 (6): 313–318. doi:10.1266/ggs.75.313. PMID 11280005.
- Sutherland B, Stewart D, Kenchington ER, Zouros E (January 1998). "The fate of paternal mitochondrial DNA in developing female mussels, Mytilus edulis: implications for the mechanism of doubly uniparental inheritance of mitochondrial DNA". Genetics. 148 (1): 341–347. PMC 1459795. PMID 9475744.
- Male and Female Mitochondrial DNA Lineages in the Blue Mussel (Mytilus edulis) Species Group Archived 2013-05-18 at the Wayback Machine by Donald T. Stewart, Carlos Saavedra, Rebecca R. Stanwood, Amy 0. Ball, and Eleftherios Zouros
- Johns DR (October 2003). "Paternal transmission of mitochondrial DNA is (fortunately) rare". Annals of Neurology. 54 (4): 422–424. doi:10.1002/ana.10771. PMID 14520651.
- Fruit flies offer DNA clue to why women live longer. BBC. 2 August 2012
- Thyagarajan B, Padua RA, Campbell C (November 1996). "Mammalian mitochondria possess homologous DNA recombination activity". The Journal of Biological Chemistry. 271 (44): 27536–27543. doi:10.1074/jbc.271.44.27536. PMID 8910339.
- Lunt DH, Hyman BC (May 1997). "Animal mitochondrial DNA recombination". Nature. 387 (6630): 247. Bibcode:1997Natur.387..247L. doi:10.1038/387247a0. PMID 9153388.
- Eyre-Walker A, Smith NH, Smith JM (March 1999). "How clonal are human mitochondria?". Proceedings. Biological Sciences. 266 (1418): 477–483. doi:10.1098/rspb.1999.0662. PMC 1689787. PMID 10189711.
- Awadalla P, Eyre-Walker A, Smith JM (December 1999). "Linkage disequilibrium and recombination in hominid mitochondrial DNA". Science. 286 (5449): 2524–2525. doi:10.1126/science.286.5449.2524. PMID 10617471.
- Cree LM, Samuels DC, de Sousa Lopes SC, Rajasimha HK, Wonnapinij P, Mann JR, Dahl HH, Chinnery PF (February 2008). "A reduction of mitochondrial DNA molecules during embryogenesis explains the rapid segregation of genotypes". Nature Genetics. 40 (2): 249–254. doi:10.1038/ng.2007.63. PMID 18223651.
- Cao L, Shitara H, Horii T, Nagao Y, Imai H, Abe K, Hara T, Hayashi J, Yonekawa H (March 2007). "The mitochondrial bottleneck occurs without reduction of mtDNA content in female mouse germ cells". Nature Genetics. 39 (3): 386–390. doi:10.1038/ng1970. PMID 17293866.
- Wai T, Teoli D, Shoubridge EA (December 2008). "The mitochondrial DNA genetic bottleneck results from replication of a subpopulation of genomes". Nature Genetics. 40 (12): 1484–1488. doi:10.1038/ng.258. PMID 19029901.
- Johnston IG, Burgstaller JP, Havlicek V, Kolbe T, Rülicke T, Brem G, Poulton J, Jones NS (June 2015). "Stochastic modelling, Bayesian inference, and new in vivo measurements elucidate the debated mtDNA bottleneck mechanism". eLife. 4: e07464. arXiv:1512.02988. doi:10.7554/eLife.07464. PMC 4486817. PMID 26035426.
- Gredilla R, Garm C, Stevnsner T (2012). "Nuclear and mitochondrial DNA repair in selected eukaryotic aging model systems". Oxid Med Cell Longev. 2012: 282438. doi:10.1155/2012/282438. PMC 3462412. PMID 23050036.
- Saki M, Prakash A (June 2017). "DNA damage related crosstalk between the nucleus and mitochondria". Free Radic. Biol. Med. 107: 216–227. doi:10.1016/j.freeradbiomed.2016.11.050. PMC 5449269. PMID 27915046.
- Leon J, Sakumi K, Castillo E, Sheng Z, Oka S, Nakabeppu Y (February 2016). "8-Oxoguanine accumulation in mitochondrial DNA causes mitochondrial dysfunction and impairs neuritogenesis in cultured adult mouse cortical neurons under oxidative conditions". Sci Rep. 6: 22086. Bibcode:2016NatSR...622086L. doi:10.1038/srep22086. PMC 4766534. PMID 26912170.
- Dahal S, Dubey S, Raghavan SC (May 2018). "Homologous recombination-mediated repair of DNA double-strand breaks operates in mammalian mitochondria". Cell. Mol. Life Sci. 75 (9): 1641–1655. doi:10.1007/s00018-017-2702-y. PMID 29116362.
- Odahara M, Inouye T, Fujita T, Hasebe M, Sekine Y (February 2007). "Involvement of mitochondrial-targeted RecA in the repair of mitochondrial DNA in the moss, Physcomitrella patens". Genes Genet. Syst. 82 (1): 43–51. doi:10.1266/ggs.82.43. PMID 17396019.
- Tadi SK, Sebastian R, Dahal S, Babu RK, Choudhary B, Raghavan SC (January 2016). "Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions". Mol. Biol. Cell. 27 (2): 223–35. doi:10.1091/mbc.E15-05-0260. PMC 4713127. PMID 26609070.
- John U, Lu Y, Wohlrab S, Groth M, Janouškovec J, Kohli GS, Mark FC, Bickmeyer U, Farhat S, Felder M, Frickenhaus S, Guillou L, Keeling PJ, Moustafa A, Porcel BM, Valentin K, Glöckner G (April 2019). "An aerobic eukaryotic parasite with functional mitochondria that likely lacks a mitochondrial genome". Science Advances. 5 (4): eaav1110. Bibcode:2019SciA....5.1110J. doi:10.1126/sciadv.aav1110. PMC 6482013. PMID 31032404.
- "Veritable powerhouses -- even without DNA: Parasitic algae from the dinoflagellate lineage have organized their genetic material in an unprecedented way". ScienceDaily.
- Castro JA, Picornell A, Ramon M (December 1998). "Mitochondrial DNA: a tool for populational genetics studies". International Microbiology. 1 (4): 327–332. PMID 10943382.
- Cann RL, Stoneking M, Wilson AC (January 1987). "Mitochondrial DNA and human evolution". Nature. 325 (6099): 31–36. Bibcode:1987Natur.325...31C. doi:10.1038/325031a0. PMID 3025745.
- Torroni A, Achilli A, Macaulay V, Richards M, Bandelt HJ (June 2006). "Harvesting the fruit of the human mtDNA tree". Trends in Genetics. 22 (6): 339–345. doi:10.1016/j.tig.2006.04.001. PMID 16678300.
- Garrigan D, Hammer MF (September 2006). "Reconstructing human origins in the genomic era". Nature Reviews Genetics. 7 (9): 669–680. doi:10.1038/nrg1941. PMID 16921345.
- Krings M, Stone A, Schmitz RW, Krainitzki H, Stoneking M, Pääbo S (July 1997). "Neandertal DNA sequences and the origin of modern humans". Cell. 90 (1): 19–30. doi:10.1016/S0092-8674(00)80310-4. hdl:11858/00-001M-0000-0025-0960-8. PMID 9230299.
- Harding RM, Fullerton SM, Griffiths RC, Bond J, Cox MJ, Schneider JA, Moulin DS, Clegg JB (April 1997). "Archaic African and Asian lineages in the genetic ancestry of modern humans". American Journal of Human Genetics. 60 (4): 772–789. PMC 1712470. PMID 9106523.
- Soares P, Ermini L, Thomson N, Mormina M, Rito T, Röhl A, Salas A, Oppenheimer S, Macaulay V, Richards MB (June 2009). "Correcting for purifying selection: an improved human mitochondrial molecular clock". American Journal of Human Genetics. 84 (6): 740–759. doi:10.1016/j.ajhg.2009.05.001. PMC 2694979. PMID 19500773.
- Nachman MW, Crowell SL (September 2000). "Estimate of the mutation rate per nucleotide in humans". Genetics. 156 (1): 297–304. PMC 1461236. PMID 10978293.
- Zeviani M, Di Donato S (October 2004). "Mitochondrial disorders". Brain. 127 (Pt 10): 2153–2172. doi:10.1093/brain/awh259. PMID 15358637.
- Taylor RW, Turnbull DM (May 2005). "Mitochondrial DNA mutations in human disease". Nature Reviews Genetics. 6 (5): 389–402. doi:10.1038/nrg1606. PMC 1762815. PMID 15861210.
- Chinnery PF, Schon EA (September 2003). "Mitochondria". Journal of Neurology, Neurosurgery, and Psychiatry. 74 (9): 1188–1199. doi:10.1136/jnnp.74.9.1188. PMC 1738655. PMID 12933917.
- Sherer TB, Betarbet R, Greenamyre JT (June 2002). "Environment, mitochondria, and Parkinson's disease". The Neuroscientist. 8 (3): 192–197. doi:10.1177/1073858402008003004. PMID 12061498.
- Gomez C, Bandez MJ, Navarro A (January 2007). "Pesticides and impairment of mitochondrial function in relation with the parkinsonian syndrome". Frontiers in Bioscience. 12: 1079–1093. doi:10.2741/2128. PMID 17127363.
- Lim YA, Rhein V, Baysang G, Meier F, Poljak A, Raftery MJ, Guilhaus M, Ittner LM, Eckert A, Götz J (April 2010). "Abeta and human amylin share a common toxicity pathway via mitochondrial dysfunction". Proteomics. 10 (8): 1621–1633. doi:10.1002/pmic.200900651. PMID 20186753.
- King, John V.; Liang, Wenguang G.; Scherpelz, Kathryn P.; Schilling, Alexander B.; Meredith, Stephen C.; Tang, Wei-Jen (2014-07-08). "Molecular basis of substrate recognition and degradation by human presequence protease". Structure (London, England: 1993). 22 (7): 996–1007. doi:10.1016/j.str.2014.05.003. ISSN 1878-4186. PMC 4128088. PMID 24931469.
- Schapira AH (July 2006). "Mitochondrial disease". Lancet. 368 (9529): 70–82. doi:10.1016/S0140-6736(06)68970-8. PMID 16815381.
- Pieczenik SR, Neustadt J (August 2007). "Mitochondrial dysfunction and molecular pathways of disease". Experimental and Molecular Pathology. 83 (1): 84–92. doi:10.1016/j.yexmp.2006.09.008. PMID 17239370.
- Bugger H, Abel ED (November 2010). "Mitochondria in the diabetic heart". Cardiovascular Research. 88 (2): 229–240. doi:10.1093/cvr/cvq239. PMC 2952534. PMID 20639213.
- Richter C, Park JW, Ames BN (September 1988). "Normal oxidative damage to mitochondrial and nuclear DNA is extensive". Proceedings of the National Academy of Sciences of the United States of America. 85 (17): 6465–6467. Bibcode:1988PNAS...85.6465R. doi:10.1073/pnas.85.17.6465. PMC 281993. PMID 3413108.
- Harman D (July 1956). "Aging: a theory based on free radical and radiation chemistry". Journal of Gerontology. 11 (3): 298–300. CiteSeerX 10.1.1.663.3809. doi:10.1093/geronj/11.3.298. PMID 13332224.
- Harman D (April 1972). "The biologic clock: the mitochondria?". Journal of the American Geriatrics Society. 20 (4): 145–147. doi:10.1111/j.1532-5415.1972.tb00787.x. PMID 5016631.
- "Mitochondria and Aging". circuitblue.co.
- Boffoli D, Scacco SC, Vergari R, Solarino G, Santacroce G, Papa S (April 1994). "Decline with age of the respiratory chain activity in human skeletal muscle". Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 1226 (1): 73–82. doi:10.1016/0925-4439(94)90061-2. PMID 8155742.
- de Grey AD (2004). "Mitochondrial mutations in mammalian aging: an over-hasty about-turn?". Rejuvenation Research. 7 (3): 171–174. doi:10.1089/rej.2004.7.171. PMID 15588517.
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM (May 2006). "High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease". Nature Genetics. 38 (5): 515–517. doi:10.1038/ng1769. PMID 16604074.
- Narcisse E (August 10, 2010). "20,000 Per Cell: Why Midi-chlorians Suck". Time. Retrieved June 19, 2016.
General

External links
- Lane, Nick (2016). The Vital Question: Energy, Evolution, and the Origins of Complex Life. WW Norton & Company. ISBN 978-0393352979.
| Wikimedia Commons has media related to Mitochondrion. |
- Mitodb.com – The mitochondrial disease database.
- Mitochondria Atlas at University of Mainz
- Mitochondria Research Portal at mitochondrial.net
- Mitochondria: Architecture dictates function at cytochemistry.net
- Mitochondria links at University of Alabama
- MIP Mitochondrial Physiology Society
- 3D structures of proteins from inner mitochondrial membrane at University of Michigan
- 3D structures of proteins associated with outer mitochondrial membrane at University of Michigan
- Mitochondrial Protein Partnership at University of Wisconsin
- MitoMiner – A mitochondrial proteomics database at MRC Mitochondrial Biology Unit
- Mitochondrion – Cell Centered Database
- Mitochondrion Reconstructed by Electron Tomography at San Diego State University
- Video Clip of Rat-liver Mitochondrion from Cryo-electron Tomography
| Authority control |
|
|---|