Population genetics
Population genetics is a subfield of genetics that deals with genetic differences within and between populations, and is a part of evolutionary biology. Studies in this branch of biology examine such phenomena as adaptation, speciation, and population structure.[1]
| Part of a series on |
| Evolutionary biology |
|---|
 |
|
Key topics |
|
Fields and applications
|
|
Social implications
|
|
| Part of a series on |
| Genetic genealogy |
|---|
| Concepts |
| Related topics |
|
Population genetics was a vital ingredient in the emergence of the modern evolutionary synthesis. Its primary founders were Sewall Wright, J. B. S. Haldane and Ronald Fisher, who also laid the foundations for the related discipline of quantitative genetics. Traditionally a highly mathematical discipline, modern population genetics encompasses theoretical, lab, and field work. Population genetic models are used both for statistical inference from DNA sequence data and for proof/disproof of concept.[2]
What sets population genetics apart today from newer, more phenotypic approaches to modelling evolution, such as evolutionary game theory and adaptive dynamics, is its emphasis on genetic phenomena as dominance, epistasis, the degree to which genetic recombination breaks up linkage disequilibrium, and the random phenomena of mutation and genetic drift. This makes it appropriate for comparison to population genomics data.
History
Population genetics began as a reconciliation of Mendelian inheritance and biostatistics models. Natural selection will only cause evolution if there is enough genetic variation in a population. Before the discovery of Mendelian genetics, one common hypothesis was blending inheritance. But with blending inheritance, genetic variance would be rapidly lost, making evolution by natural or sexual selection implausible. The Hardy–Weinberg principle provides the solution to how variation is maintained in a population with Mendelian inheritance. According to this principle, the frequencies of alleles (variations in a gene) will remain constant in the absence of selection, mutation, migration and genetic drift.[3]


The next key step was the work of the British biologist and statistician Ronald Fisher. In a series of papers starting in 1918 and culminating in his 1930 book The Genetical Theory of Natural Selection, Fisher showed that the continuous variation measured by the biometricians could be produced by the combined action of many discrete genes, and that natural selection could change allele frequencies in a population, resulting in evolution. In a series of papers beginning in 1924, another British geneticist, J. B. S. Haldane, worked out the mathematics of allele frequency change at a single gene locus under a broad range of conditions. Haldane also applied statistical analysis to real-world examples of natural selection, such as peppered moth evolution and industrial melanism, and showed that selection coefficients could be larger than Fisher assumed, leading to more rapid adaptive evolution as a camouflage strategy following increased pollution.[4][5]
The American biologist Sewall Wright, who had a background in animal breeding experiments, focused on combinations of interacting genes, and the effects of inbreeding on small, relatively isolated populations that exhibited genetic drift. In 1932 Wright introduced the concept of an adaptive landscape and argued that genetic drift and inbreeding could drive a small, isolated sub-population away from an adaptive peak, allowing natural selection to drive it towards different adaptive peaks.
The work of Fisher, Haldane and Wright founded the discipline of population genetics. This integrated natural selection with Mendelian genetics, which was the critical first step in developing a unified theory of how evolution worked.[4][5] John Maynard Smith was Haldane's pupil, whilst W. D. Hamilton was heavily influenced by the writings of Fisher. The American George R. Price worked with both Hamilton and Maynard Smith. American Richard Lewontin and Japanese Motoo Kimura were heavily influenced by Wright and Haldane.
Modern synthesis
The mathematics of population genetics were originally developed as the beginning of the modern synthesis. Authors such as Beatty[6] have asserted that population genetics defines the core of the modern synthesis. For the first few decades of the 20th century, most field naturalists continued to believe that Lamarckism and orthogenesis provided the best explanation for the complexity they observed in the living world.[7] During the modern synthesis, these ideas were purged, and only evolutionary causes that could be expressed in the mathematical framework of population genetics were retained.[8] Consensus was reached as to which evolutionary factors might influence evolution, but not as to the relative importance of the various factors.[8]
Theodosius Dobzhansky, a postdoctoral worker in T. H. Morgan's lab, had been influenced by the work on genetic diversity by Russian geneticists such as Sergei Chetverikov. He helped to bridge the divide between the foundations of microevolution developed by the population geneticists and the patterns of macroevolution observed by field biologists, with his 1937 book Genetics and the Origin of Species. Dobzhansky examined the genetic diversity of wild populations and showed that, contrary to the assumptions of the population geneticists, these populations had large amounts of genetic diversity, with marked differences between sub-populations. The book also took the highly mathematical work of the population geneticists and put it into a more accessible form. Many more biologists were influenced by population genetics via Dobzhansky than were able to read the highly mathematical works in the original.[9]
In Great Britain E. B. Ford, the pioneer of ecological genetics, continued throughout the 1930s and 1940s to empirically demonstrate the power of selection due to ecological factors including the ability to maintain genetic diversity through genetic polymorphisms such as human blood types. Ford's work, in collaboration with Fisher, contributed to a shift in emphasis during the course of the modern synthesis towards natural selection as the dominant force.[4][5][10][11]
Neutral theory and origin-fixation dynamics
The original, modern synthesis view of population genetics assumes that mutations provide ample raw material, and focuses only on the change in frequency of alleles within populations.[12] The main processes influencing allele frequencies are natural selection, genetic drift, gene flow and recurrent mutation. Fisher and Wright had some fundamental disagreements about the relative roles of selection and drift.[13]
The availability of molecular data on all genetic differences led to the neutral theory of molecular evolution. In this view, many mutations are deleterious and so never observed, and most of the remainder are neutral, i.e. are not under selection. With the fate of each neutral mutation left to chance (genetic drift), the direction of evolutionary change is driven by which mutations occur, and so cannot be captured by models of change in the frequency of (existing) alleles alone.[12][14]
The origin-fixation view of population genetics generalizes this approach beyond strictly neutral mutations, and sees the rate at which a particular change happens as the product of the mutation rate and the fixation probability.[12]
Four processes
Selection
Natural selection, which includes sexual selection, is the fact that some traits make it more likely for an organism to survive and reproduce. Population genetics describes natural selection by defining fitness as a propensity or probability of survival and reproduction in a particular environment. The fitness is normally given by the symbol w=1-s where s is the selection coefficient. Natural selection acts on phenotypes, so population genetic models assume relatively simple relationships to predict the phenotype and hence fitness from the allele at one or a small number of loci. In this way, natural selection converts differences in the fitness of individuals with different phenotypes into changes in allele frequency in a population over successive generations.
Before the advent of population genetics, many biologists doubted that small differences in fitness were sufficient to make a large difference to evolution.[9] Population geneticists addressed this concern in part by comparing selection to genetic drift. Selection can overcome genetic drift when s is greater than 1 divided by the effective population size. When this criterion is met, the probability that a new advantageous mutant becomes fixed is approximately equal to 2s.[15][16] The time until fixation of such an allele depends little on genetic drift, and is approximately proportional to log(sN)/s.[17]
Dominance
Dominance means that the phenotypic and/or fitness effect of one allele at a locus depends on which allele is present in the second copy for that locus. Consider three genotypes at one locus, with the following fitness values[18]
| - | Genotype: | A1A1 | A1A2 | A2A2 | - | Relative fitness: | 1 | 1-hs | 1-s |
s is the selection coefficient and h is the dominance coefficient. The value of h yields the following information:
| - | h=0 | A1 dominant, A2 recessive | - | h=1 | A2 dominant, A1 recessive | - | 0<h<1 | incomplete dominance | - | h<0 | overdominance | - | h>1 | Underdominance |
Epistasis
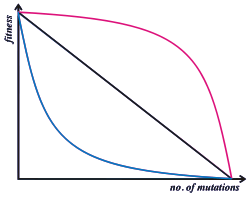
Epistasis means that the phenotypic and/or fitness effect of an allele at one locus depends on which alleles are present at other loci. Selection does not act on a single locus, but on a phenotype that arises through development from a complete genotype.[19] However, many population genetics models of sexual species are "single locus" models, where the fitness of an individual is calculated as the product of the contributions from each of its loci—effectively assuming no epistasis.
In fact, the genotype to fitness landscape is more complex. Population genetics must either model this complexity in detail, or capture it by some simpler average rule. Empirically, beneficial mutations tend to have a smaller fitness benefit when added to a genetic background that already has high fitness: this is known as diminishing returns epistasis.[20] When deleterious mutations also have a smaller fitness effect on high fitness backgrounds, this is known as "synergistic epistasis". However, the effect of deleterious mutations tends on average to be very close to multiplicative, or can even show the opposite pattern, known as "antagonistic epistasis".[21]
Synergistic epistasis is central to some theories of the purging of mutation load[22] and to the evolution of sexual reproduction.
Mutation
.jpg)
Mutation is the ultimate source of genetic variation in the form of new alleles. In addition, mutation may influence the direction of evolution when there is mutation bias, i.e. different probabilities for different mutations to occur. For example, recurrent mutation that tends to be in the opposite direction to selection can lead to mutation–selection balance. At the molecular level, if mutation from G to A happens more often than mutation from A to G, then genotypes with A will tend to evolve.[23] Different insertion vs. deletion mutation biases in different taxa can lead to the evolution of different genome sizes.[24][25] Developmental or mutational biases have also been observed in morphological evolution.[26][27] For example, according to the phenotype-first theory of evolution, mutations can eventually cause the genetic assimilation of traits that were previously induced by the environment.[28][29]
Mutation bias effects are superimposed on other processes. If selection would favor either one out of two mutations, but there is no extra advantage to having both, then the mutation that occurs the most frequently is the one that is most likely to become fixed in a population.[30][31]
Mutation can have no effect, alter the product of a gene, or prevent the gene from functioning. Studies in the fly Drosophila melanogaster suggest that if a mutation changes a protein produced by a gene, this will probably be harmful, with about 70 percent of these mutations having damaging effects, and the remainder being either neutral or weakly beneficial.[32] Most loss of function mutations are selected against. But when selection is weak, mutation bias towards loss of function can affect evolution.[33] For example, pigments are no longer useful when animals live in the darkness of caves, and tend to be lost.[34] This kind of loss of function can occur because of mutation bias, and/or because the function had a cost, and once the benefit of the function disappeared, natural selection leads to the loss. Loss of sporulation ability in a bacterium during laboratory evolution appears to have been caused by mutation bias, rather than natural selection against the cost of maintaining sporulation ability.[35] When there is no selection for loss of function, the speed at which loss evolves depends more on the mutation rate than it does on the effective population size,[36] indicating that it is driven more by mutation bias than by genetic drift.
Mutations can involve large sections of DNA becoming duplicated, usually through genetic recombination.[37] This leads to copy-number variation within a population. Duplications are a major source of raw material for evolving new genes.[38] Other types of mutation occasionally create new genes from previously noncoding DNA.[39][40]
Genetic drift
Genetic drift is a change in allele frequencies caused by random sampling.[41] That is, the alleles in the offspring are a random sample of those in the parents.[42] Genetic drift may cause gene variants to disappear completely, and thereby reduce genetic variability. In contrast to natural selection, which makes gene variants more common or less common depending on their reproductive success,[43] the changes due to genetic drift are not driven by environmental or adaptive pressures, and are equally likely to make an allele more common as less common.
The effect of genetic drift is larger for alleles present in few copies than when an allele is present in many copies. The population genetics of genetic drift are described using either branching processes or a diffusion equation describing changes in allele frequency.[44] These approaches are usually applied to the Wright-Fisher and Moran models of population genetics. Assuming genetic drift is the only evolutionary force acting on an allele, after t generations in many replicated populations, starting with allele frequencies of p and q, the variance in allele frequency across those populations is

Ronald Fisher held the view that genetic drift plays at the most a minor role in evolution, and this remained the dominant view for several decades. No population genetics perspective have ever given genetic drift a central role by itself, but some have made genetic drift important in combination with another non-selective force. The shifting balance theory of Sewall Wright held that the combination of population structure and genetic drift was important. Motoo Kimura's neutral theory of molecular evolution claims that most genetic differences within and between populations are caused by the combination of neutral mutations and genetic drift.[46]
The role of genetic drift by means of sampling error in evolution has been criticized by John H Gillespie[47] and Will Provine,[48] who argue that selection on linked sites is a more important stochastic force, doing the work traditionally ascribed to genetic drift by means of sampling error. The mathematical properties of genetic draft are different from those of genetic drift.[49] The direction of the random change in allele frequency is autocorrelated across generations.[41]
Gene flow
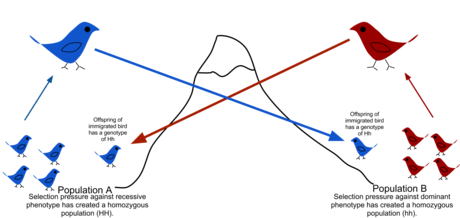

Because of physical barriers to migration, along with the limited tendency for individuals to move or spread (vagility), and tendency to remain or come back to natal place (philopatry), natural populations rarely all interbreed as may be assumed in theoretical random models (panmixy).[50] There is usually a geographic range within which individuals are more closely related to one another than those randomly selected from the general population. This is described as the extent to which a population is genetically structured.[51] Genetic structuring can be caused by migration due to historical climate change, species range expansion or current availability of habitat. Gene flow is hindered by mountain ranges, oceans and deserts or even man-made structures such as the Great Wall of China, which has hindered the flow of plant genes.[52]
Gene flow is the exchange of genes between populations or species, breaking down the structure. Examples of gene flow within a species include the migration and then breeding of organisms, or the exchange of pollen. Gene transfer between species includes the formation of hybrid organisms and horizontal gene transfer. Population genetic models can be used to identify which populations show significant genetic isolation from one another, and to reconstruct their history.[53]
Subjecting a population to isolation leads to inbreeding depression. Migration into a population can introduce new genetic variants,[54] potentially contributing to evolutionary rescue. If a significant proportion of individuals or gametes migrate, it can also change allele frequencies, e.g. giving rise to migration load.[55]
In the presence of gene flow, other barriers to hybridization between two diverging populations of an outcrossing species are required for the populations to become new species.
Horizontal gene transfer
Horizontal gene transfer is the transfer of genetic material from one organism to another organism that is not its offspring; this is most common among prokaryotes.[56] In medicine, this contributes to the spread of antibiotic resistance, as when one bacteria acquires resistance genes it can rapidly transfer them to other species.[57] Horizontal transfer of genes from bacteria to eukaryotes such as the yeast Saccharomyces cerevisiae and the adzuki bean beetle Callosobruchus chinensis may also have occurred.[58][59] An example of larger-scale transfers are the eukaryotic bdelloid rotifers, which appear to have received a range of genes from bacteria, fungi, and plants.[60] Viruses can also carry DNA between organisms, allowing transfer of genes even across biological domains.[61] Large-scale gene transfer has also occurred between the ancestors of eukaryotic cells and prokaryotes, during the acquisition of chloroplasts and mitochondria.[62]
Linkage
If all genes are in linkage equilibrium, the effect of an allele at one locus can be averaged across the gene pool at other loci. In reality, one allele is frequently found in linkage disequilibrium with genes at other loci, especially with genes located nearby on the same chromosome. Recombination breaks up this linkage disequilibrium too slowly to avoid genetic hitchhiking, where an allele at one locus rises to high frequency because it is linked to an allele under selection at a nearby locus. Linkage also slows down the rate of adaptation, even in sexual populations.[63][64][65] The effect of linkage disequilibrium in slowing down the rate of adaptive evolution arises from a combination of the Hill–Robertson effect (delays in bringing beneficial mutations together) and background selection (delays in separating beneficial mutations from deleterious hitchhikers).
Linkage is a problem for population genetic models that treat one gene locus at a time. It can, however, be exploited as a method for detecting the action of natural selection via selective sweeps.
In the extreme case of an asexual population, linkage is complete, and population genetic equations can be derived and solved in terms of a travelling wave of genotype frequencies along a simple fitness landscape.[66] Most microbes, such as bacteria, are asexual. The population genetics of their adaptation have two contrasting regimes. When the product of the beneficial mutation rate and population size is small, asexual populations follow a "successional regime" of origin-fixation dynamics, with adaptation rate strongly dependent on this product. When the product is much larger, asexual populations follow a "concurrent mutations" regime with adaptation rate less dependent on the product, characterized by clonal interference and the appearance of a new beneficial mutation before the last one has fixed.
Applications
Explaining levels of genetic variation
Neutral theory predicts that the level of nucleotide diversity in a population will be proportional to the product of the population size and the neutral mutation rate. The fact that levels of genetic diversity vary much less than population sizes do is known as the "paradox of variation".[67] While high levels of genetic diversity were one of the original arguments in favor of neutral theory, the paradox of variation has been one of the strongest arguments against neutral theory.
It is clear that levels of genetic diversity vary greatly within a species as a function of local recombination rate, due to both genetic hitchhiking and background selection. Most current solutions to the paradox of variation invoke some level of selection at linked sites.[68] For example, one analysis suggests that larger populations have more selective sweeps, which remove more neutral genetic diversity.[69] A negative correlation between mutation rate and population size may also contribute.[70]
Life history affects genetic diversity more than population history does, e.g. r-strategists have more genetic diversity.[68]
Detecting selection
Population genetics models are used to infer which genes are undergoing selection. One common approach is to look for regions of high linkage disequilibrium and low genetic variance along the chromosome, to detect recent selective sweeps.
A second common approach is the McDonald–Kreitman test. The McDonald–Kreitman test compares the amount of variation within a species (polymorphism) to the divergence between species (substitutions) at two types of sites, one assumed to be neutral. Typically, synonymous sites are assumed to be neutral.[71] Genes undergoing positive selection have an excess of divergent sites relative to polymorphic sites. The test can also be use to obtain a genome-wide estimate of the proportion of substitutions that are fixed by positive selection, α.[72][73] According to the neutral theory of molecular evolution, this number should be near zero. High numbers have therefore been interpreted as a genome-wide falsification of neutral theory.[74]
Demographic inference
The simplest test for population structure in a sexually reproducing, diploid species, is to see whether genotype frequencies follow Hardy-Weinberg proportions as a function of allele frequencies. For example, in the simplest case of a single locus with two alleles denoted A and a at frequencies p and q, random mating predicts freq(AA) = p2 for the AA homozygotes, freq(aa) = q2 for the aa homozygotes, and freq(Aa) = 2pq for the heterozygotes. In the absence of population structure, Hardy-Weinberg proportions are reached within 1-2 generations of random mating. More typically, there is an excess of homozygotes, indicative of population structure. The extent of this excess can be quantified as the inbreeding coefficient, F.
Individuals can be clustered into K subpopulations.[75][76] The degree of population structure can then be calculated using FST, which is a measure of the proportion of genetic variance that can be explained by population structure. Genetic population structure can then be related to geographic structure, and genetic admixture can be detected.
Coalescent theory relates genetic diversity in a sample to demographic history of the population from which it was taken. It normally assumes neutrality, and so sequences from more neutrally-evolving portions of genomes are therefore selected for such analyses. It can be used to infer the relationships between species (phylogenetics), as well as the population structure, demographic history (e.g. population bottlenecks, population growth), biological dispersal, source–sink dynamics[77] and introgression within a species.
Another approach to demographic inference relies on the allele frequency spectrum.[78]
Evolution of genetic systems
By assuming that there are loci that control the genetic system itself, population genetic models are created to describe the evolution of dominance and other forms of robustness, the evolution of sexual reproduction and recombination rates, the evolution of mutation rates, the evolution of evolutionary capacitors, the evolution of costly signalling traits, the evolution of ageing, and the evolution of co-operation. For example, most mutations are deleterious, so the optimal mutation rate for a species may be a trade-off between the damage from a high deleterious mutation rate and the metabolic costs of maintaining systems to reduce the mutation rate, such as DNA repair enzymes.[79]
One important aspect of such models is that selection is only strong enough to purge deleterious mutations and hence overpower mutational bias towards degradation if the selection coefficient s is greater than the inverse of the effective population size. This is known as the drift barrier and is related to the nearly neutral theory of molecular evolution. Drift barrier theory predicts that species with large effective population sizes will have highly streamlined, efficient genetic systems, while those with small population sizes will have bloated and complex genomes containing for example introns and transposable elements.[80] However, somewhat paradoxically, species with large population sizes might be so tolerant to the consequences of certain types of errors that they evolve higher error rates, e.g. in transcription and translation, than small populations.[81]
See also
References
- "Population genetics - Latest research and news | Nature". www.nature.com. Retrieved 2018-01-29.
- Servedio, Maria R.; Brandvain, Yaniv; Dhole, Sumit; Fitzpatrick, Courtney L.; Goldberg, Emma E.; Stern, Caitlin A.; Van Cleve, Jeremy; Yeh, D. Justin (9 December 2014). "Not Just a Theory—The Utility of Mathematical Models in Evolutionary Biology". PLOS Biology. 12 (12): e1002017. doi:10.1371/journal.pbio.1002017. PMC 4260780. PMID 25489940.
- Ewens W.J. (2004). Mathematical Population Genetics (2nd ed.). Springer-Verlag, New York. ISBN 978-0-387-20191-7.
- Bowler, Peter J. (2003). Evolution : the history of an idea (3rd ed.). Berkeley: University of California Press. pp. 325–339. ISBN 978-0-520-23693-6.
- Larson, Edward J. (2004). Evolution : the remarkable history of a scientific theory (Modern Library ed.). New York: Modern Library. pp. 221–243. ISBN 978-0-679-64288-6.
- Beatty, John (1986). "The Synthesis and the Synthetic Theory". Integrating Scientific Disciplines. Science and Philosophy. 2. Springer Netherlands. pp. 125–135. doi:10.1007/978-94-010-9435-1_7. ISBN 9789024733422.
- Mayr, edited by Ernst; Mayer, William B. Provine; with a new preface by Ernst (1998). The Evolutionary synthesis : perspectives on the unification of biology ([New ed]. ed.). Cambridge, Massachusetts: Harvard University Press. pp. 295–298. ISBN 9780674272262.CS1 maint: extra text: authors list (link)
- Provine, W. B. (1988). "Progress in evolution and meaning in life". Evolutionary progress. University of Chicago Press. pp. 49–79.
- Provine, William B. (1978). "The role of mathematical population geneticists in the evolutionary synthesis of the 1930s and 1940s". Studies of the History of Biology. 2: 167–192. PMID 11610409.
- Mayr, Ernst (1988). Toward a New Philosophy of Biology: Observations of an Evolutionist. Cambridge, Massachusetts: Belknap Press of Harvard University Press. p. 402. ISBN 978-0-674-89665-9.CS1 maint: ref=harv (link)
- Mayr, edited by Ernst; Mayer, William B. Provine; with a new preface by Ernst (1998). The Evolutionary synthesis : perspectives on the unification of biology ([New ed]. ed.). Cambridge, Massachusetts: Harvard University Press. pp. 338–341. ISBN 9780674272262.CS1 maint: extra text: authors list (link)
- McCandlish, David M.; Stoltzfus, Arlin (September 2014). "Modeling Evolution Using the Probability of Fixation: History and Implications". The Quarterly Review of Biology. 89 (3): 225–252. doi:10.1086/677571. PMID 25195318.
- Wright and Fisher on Inbreeding and Random Drift by James F. Crow, published in Genetics Published 2010
- Casillas, Sònia; Barbadilla, Antonio (2017). "Molecular Population Genetics". Genetics. 205 (3): 1003–1035. doi:10.1534/genetics.116.196493. PMC 5340319. PMID 28270526.
- JBS Haldane (1927). "A Mathematical Theory of Natural and Artificial Selection, Part V: Selection and Mutation". Mathematical Proceedings of the Cambridge Philosophical Society. 23 (7): 838–844. Bibcode:1927PCPS...23..838H. doi:10.1017/S0305004100015644.
- Orr, H. A. (2010). "The population genetics of beneficial mutations". Philosophical Transactions of the Royal Society B: Biological Sciences. 365 (1544): 1195–1201. doi:10.1098/rstb.2009.0282. PMC 2871816. PMID 20308094.
- Hermisson J, Pennings PS; Pennings (2005). "Soft sweeps: molecular population genetics of adaptation from standing genetic variation". Genetics. 169 (4): 2335–2352. doi:10.1534/genetics.104.036947. PMC 1449620. PMID 15716498.
- Gillespie, John (2004). Population Genetics: A Concise Guide (2nd ed.). Johns Hopkins University Press. ISBN 978-0-8018-8008-7.
- Miko, I. (2008). "Epistasis: Gene interaction and phenotype effects". Nature Education. 1 (1): 197.
- Berger, D.; Postma, E. (13 October 2014). "Biased Estimates of Diminishing-Returns Epistasis? Empirical Evidence Revisited". Genetics. 198 (4): 1417–1420. doi:10.1534/genetics.114.169870. PMC 4256761. PMID 25313131.
- Kouyos, Roger D.; Silander, Olin K.; Bonhoeffer, Sebastian (June 2007). "Epistasis between deleterious mutations and the evolution of recombination". Trends in Ecology & Evolution. 22 (6): 308–315. doi:10.1016/j.tree.2007.02.014. PMID 17337087.
- Crow, J. F. (5 August 1997). "The high spontaneous mutation rate: is it a health risk?". Proceedings of the National Academy of Sciences of the United States of America. 94 (16): 8380–8386. Bibcode:1997PNAS...94.8380C. doi:10.1073/pnas.94.16.8380. PMC 33757. PMID 9237985.
- Smith N.G.C., Webster M.T., Ellegren, H.; Webster; Ellegren (2002). "Deterministic Mutation Rate Variation in the Human Genome". Genome Research. 12 (9): 1350–1356. doi:10.1101/gr.220502. PMC 186654. PMID 12213772.CS1 maint: multiple names: authors list (link)
- Petrov DA, Sangster TA, Johnston JS, Hartl DL, Shaw KL; Sangster; Johnston; Hartl; Shaw (2000). "Evidence for DNA loss as a determinant of genome size". Science. 287 (5455): 1060–1062. Bibcode:2000Sci...287.1060P. doi:10.1126/science.287.5455.1060. PMID 10669421.CS1 maint: multiple names: authors list (link)
- Petrov DA (2002). "DNA loss and evolution of genome size in Drosophila". Genetica. 115 (1): 81–91. doi:10.1023/A:1016076215168. PMID 12188050.
- Kiontke K, Barriere A, Kolotuev I, Podbilewicz B, Sommer R, Fitch DHA, Felix MA; Barrière; Kolotuev; Podbilewicz; Sommer; Fitch; Félix (2007). "Trends, stasis, and drift in the evolution of nematode vulva development". Current Biology. 17 (22): 1925–1937. doi:10.1016/j.cub.2007.10.061. PMID 18024125.CS1 maint: multiple names: authors list (link)
- Braendle C, Baer CF, Felix MA; Baer; Félix (2010). Barsh, Gregory S (ed.). "Bias and Evolution of the Mutationally Accessible Phenotypic Space in a Developmental System". PLOS Genetics. 6 (3): e1000877. doi:10.1371/journal.pgen.1000877. PMC 2837400. PMID 20300655.CS1 maint: multiple names: authors list (link)
- Palmer, RA (2004). "Symmetry breaking and the evolution of development". Science. 306 (5697): 828–833. Bibcode:2004Sci...306..828P. CiteSeerX 10.1.1.631.4256. doi:10.1126/science.1103707. PMID 15514148.
- West-Eberhard, M-J. (2003). Developmental plasticity and evolution. New York: Oxford University Press. ISBN 978-0-19-512235-0.
- Stoltzfus, A & Yampolsky, L.Y. (2009). "Climbing Mount Probable: Mutation as a Cause of Nonrandomness in Evolution". J Hered. 100 (5): 637–647. doi:10.1093/jhered/esp048. PMID 19625453.
- Yampolsky, L.Y. & Stoltzfus, A (2001). "Bias in the introduction of variation as an orienting factor in evolution". Evol Dev. 3 (2): 73–83. doi:10.1046/j.1525-142x.2001.003002073.x. PMID 11341676.
- Sawyer SA, Parsch J, Zhang Z, Hartl DL; Parsch; Zhang; Hartl (2007). "Prevalence of positive selection among nearly neutral amino acid replacements in Drosophila". Proc. Natl. Acad. Sci. U.S.A. 104 (16): 6504–10. Bibcode:2007PNAS..104.6504S. doi:10.1073/pnas.0701572104. PMC 1871816. PMID 17409186.CS1 maint: multiple names: authors list (link) CS1 maint: ref=harv (link)
- Haldane, JBS (1933). "The Part Played by Recurrent Mutation in Evolution". American Naturalist. 67 (708): 5–19. doi:10.1086/280465. JSTOR 2457127.
- Protas, Meredith; Conrad, M; Gross, JB; Tabin, C; Borowsky, R (2007). "Regressive evolution in the Mexican cave tetra, Astyanax mexicanus". Current Biology. 17 (5): 452–454. doi:10.1016/j.cub.2007.01.051. PMC 2570642. PMID 17306543.CS1 maint: ref=harv (link)
- Maughan H, Masel J, Birky WC, Nicholson WL; Masel; Birky Jr; Nicholson (2007). "The roles of mutation accumulation and selection in loss of sporulation in experimental populations of Bacillus subtilis". Genetics. 177 (2): 937–948. doi:10.1534/genetics.107.075663. PMC 2034656. PMID 17720926.CS1 maint: multiple names: authors list (link)
- Masel J, King OD, Maughan H; King; Maughan (2007). "The loss of adaptive plasticity during long periods of environmental stasis". American Naturalist. 169 (1): 38–46. doi:10.1086/510212. PMC 1766558. PMID 17206583.CS1 maint: multiple names: authors list (link)
- Hastings, P J; Lupski, JR; Rosenberg, SM; Ira, G (2009). "Mechanisms of change in gene copy number". Nature Reviews Genetics. 10 (8): 551–564. doi:10.1038/nrg2593. PMC 2864001. PMID 19597530.CS1 maint: ref=harv (link)
- Long M, Betrán E, Thornton K, Wang W; Betrán; Thornton; Wang (November 2003). "The origin of new genes: glimpses from the young and old". Nat. Rev. Genet. 4 (11): 865–75. doi:10.1038/nrg1204. PMID 14634634.CS1 maint: multiple names: authors list (link) CS1 maint: ref=harv (link)
- Liu N, Okamura K, Tyler DM; Okamura; Tyler; Phillips; Chung; Lai (2008). "The evolution and functional diversification of animal microRNA genes". Cell Res. 18 (10): 985–96. doi:10.1038/cr.2008.278. PMC 2712117. PMID 18711447.CS1 maint: multiple names: authors list (link) CS1 maint: ref=harv (link)
- McLysaght, Aoife; Hurst, Laurence D. (25 July 2016). "Open questions in the study of de novo genes: what, how and why". Nature Reviews Genetics. 17 (9): 567–578. doi:10.1038/nrg.2016.78. PMID 27452112.
- Masel, J. (2011). "Genetic drift". Current Biology. 21 (20): R837–R838. doi:10.1016/j.cub.2011.08.007. PMID 22032182.
- Futuyma, Douglas (1998). Evolutionary Biology. Sinauer Associates. p. Glossary. ISBN 978-0-87893-189-7.
- Avers, Charlotte (1989). "Process and Pattern in Evolution". Oxford University Press. Cite journal requires
|journal=(help)CS1 maint: ref=harv (link) - Wahl L.M. (2011). "Fixation when N and s Vary: Classic Approaches Give Elegant New Results". Genetics. 188 (4): 783–785. doi:10.1534/genetics.111.131748. PMC 3176088. PMID 21828279.
- Nicholas H. Barton; Derek E. G. Briggs; Jonathan A. Eisen; David B. Goldstein; Nipam H. Patel (2007). Evolution. Cold Spring Harbor Laboratory Press. p. 417. ISBN 978-0-87969-684-9.
- Futuyma, Douglas (1998). Evolutionary Biology. Sinauer Associates. p. 320. ISBN 978-0-87893-189-7.
- Gillespie, J.H. (2000). "Genetic Drift in an Infinite Population: The Pseudohitchhiking Model". Genetics. 155 (2): 909–919. PMC 1461093. PMID 10835409.
- Provine, William B. The "Random Genetic Drift" Fallacy. CreateSpace.
- Neher, Richard A.; Shraiman, Boris I. (August 2011). "Genetic Draft and Quasi-Neutrality in Large Facultatively Sexual Populations". Genetics. 188 (4): 975–996. doi:10.1534/genetics.111.128876. ISSN 0016-6731. PMC 3176096. PMID 21625002.
- Buston, P. M.; Pilkington, J. G.; et al. (2007). "Are clownfish groups composed of close relatives? An analysis of microsatellite DNA vraiation in Amphiprion percula". Molecular Ecology. 12 (3): 733–742. doi:10.1046/j.1365-294X.2003.01762.x. PMID 12675828.
- Repaci, V.; Stow, A.J.; Briscoe, D.A. (2007). "Fine-scale genetic structure, co-founding and multiple mating in the Australian allodapine bee (Ramphocinclus brachyurus)". Journal of Zoology. 270 (4): 687–691. doi:10.1111/j.1469-7998.2006.00191.x.CS1 maint: ref=harv (link)
- Su, H.; Qu, L.; He, K., Zhang, Z.; Wang, J.; Chen, Z.; Gu, H.; Qu; He; Zhang; Wang; Chen; Gu (2003). "The Great Wall of China: a physical barrier to gene flow?". Heredity. 90 (3): 212–9. doi:10.1038/sj.hdy.6800237. PMID 12634804.CS1 maint: multiple names: authors list (link) CS1 maint: ref=harv (link)
- Gravel, S., S. (2012). "Population Genetics Models of Local Ancestry". Genetics. 1202 (2): 607–619. arXiv:1202.4811. Bibcode:2012arXiv1202.4811G. doi:10.1534/genetics.112.139808. PMC 3374321. PMID 22491189.
- Morjan, C.; Rieseberg, L.; Rieseberg (2004). "How species evolve collectively: implications of gene flow and selection for the spread of advantageous alleles". Mol. Ecol. 13 (6): 1341–56. doi:10.1111/j.1365-294X.2004.02164.x. PMC 2600545. PMID 15140081.CS1 maint: multiple names: authors list (link) CS1 maint: ref=harv (link)
- Bolnick, Daniel I.; Nosil, Patrik (September 2007). "Natural Selection in Populations Subject to a Migration Load". Evolution. 61 (9): 2229–2243. doi:10.1111/j.1558-5646.2007.00179.x. PMID 17767592.
- Boucher, Y.; Douady, C.J.; Papke, R.T.; Walsh, D.A.; Boudreau, M.E.; Nesbo, C.L.; Case, R.J.; Doolittle, W.F.; Douady; Papke; Walsh; Boudreau; Nesbø; Case; Doolittle (2003). "Lateral gene transfer and the origins of prokaryotic groups". Annu Rev Genet. 37: 283–328. doi:10.1146/annurev.genet.37.050503.084247. PMID 14616063.CS1 maint: multiple names: authors list (link)
- Walsh T (2006). "Combinatorial genetic evolution of multiresistance". Curr. Opin. Microbiol. 9 (5): 476–82. doi:10.1016/j.mib.2006.08.009. PMID 16942901.
- Kondo N, Nikoh N, Ijichi N, Shimada M, Fukatsu T; Nikoh; Ijichi; Shimada; Fukatsu (2002). "Genome fragment of Wolbachia endosymbiont transferred to X chromosome of host insect". PNAS. 99 (22): 14280–5. Bibcode:2002PNAS...9914280K. doi:10.1073/pnas.222228199. PMC 137875. PMID 12386340.CS1 maint: multiple names: authors list (link)
- Sprague G (1991). "Genetic exchange between kingdoms". Curr. Opin. Genet. Dev. 1 (4): 530–3. doi:10.1016/S0959-437X(05)80203-5. PMID 1822285.
- Gladyshev EA, Meselson M, Arkhipova IR; Meselson; Arkhipova (May 2008). "Massive horizontal gene transfer in bdelloid rotifers". Science. 320 (5880): 1210–3. Bibcode:2008Sci...320.1210G. doi:10.1126/science.1156407. PMID 18511688.CS1 maint: multiple names: authors list (link)
- Baldo A, McClure M; McClure (1 September 1999). "Evolution and horizontal transfer of dUTPase-encoding genes in viruses and their hosts". J. Virol. 73 (9): 7710–21. doi:10.1128/JVI.73.9.7710-7721.1999. PMC 104298. PMID 10438861.
- Poole A, Penny D; Penny (2007). "Evaluating hypotheses for the origin of eukaryotes". BioEssays. 29 (1): 74–84. doi:10.1002/bies.20516. PMID 17187354.
- Weissman, D. B.; Hallatschek, O. (15 January 2014). "The Rate of Adaptation in Large Sexual Populations with Linear Chromosomes". Genetics. 196 (4): 1167–1183. doi:10.1534/genetics.113.160705. PMC 3982688. PMID 24429280.
- Weissman, Daniel B.; Barton, Nicholas H.; McVean, Gil (7 June 2012). "Limits to the Rate of Adaptive Substitution in Sexual Populations". PLOS Genetics. 8 (6): e1002740. doi:10.1371/journal.pgen.1002740. PMC 3369949. PMID 22685419.
- Neher, R. A.; Shraiman, B. I.; Fisher, D. S. (30 November 2009). "Rate of Adaptation in Large Sexual Populations". Genetics. 184 (2): 467–481. arXiv:1108.3464. doi:10.1534/genetics.109.109009. PMC 2828726. PMID 19948891.
- Michael M. Desai, Daniel S. Fisher; Fisher (2007). "Beneficial Mutation Selection Balance and the Effect of Linkage on Positive Selection". Genetics. 176 (3): 1759–1798. doi:10.1534/genetics.106.067678. PMC 1931526. PMID 17483432.
- Lewontin, [by] R. C. (1973). The genetic basis of evolutionary change ([4th printing.] ed.). New York: Columbia University Press. ISBN 978-0231033923.
- Ellegren, Hans; Galtier, Nicolas (6 June 2016). "Determinants of genetic diversity". Nature Reviews Genetics. 17 (7): 422–433. doi:10.1038/nrg.2016.58. PMID 27265362.
- Corbett-Detig, Russell B.; Hartl, Daniel L.; Sackton, Timothy B.; Barton, Nick H. (10 April 2015). "Natural Selection Constrains Neutral Diversity across A Wide Range of Species". PLOS Biology. 13 (4): e1002112. doi:10.1371/journal.pbio.1002112. PMC 4393120. PMID 25859758.
- Sung, W.; Ackerman, M. S.; Miller, S. F.; Doak, T. G.; Lynch, M. (17 October 2012). "Drift-barrier hypothesis and mutation-rate evolution" (PDF). Proceedings of the National Academy of Sciences. 109 (45): 18488–18492. Bibcode:2012PNAS..10918488S. doi:10.1073/pnas.1216223109. PMC 3494944. PMID 23077252.
- Charlesworth, J. Eyre-Walker (2008). "The McDonald–Kreitman Test and Slightly Deleterious Mutations". Molecular Biology and Evolution. 25 (6): 1007–1015. doi:10.1093/molbev/msn005. PMID 18195052.
- Eyre-Walker, A (2006). "The genomic rate of adaptive evolution" (PDF). Trends in Ecology and Evolution. 21 (10): 569–575. doi:10.1016/j.tree.2006.06.015. PMID 16820244.
- Smith, N. G. C.; Eyre-Walker, A. (2002). "Adaptive protein evolution in Drosophila". Nature. 415 (6875): 1022–1024. doi:10.1038/4151022a. PMID 11875568.
- Hahn, M.W. (2008). "Toward a selection theory of molecular evolution". Evolution. 62 (2): 255–265. doi:10.1111/j.1558-5646.2007.00308.x. PMID 18302709.
- Pritchard, J K; Stephens, M; Donnelly, P (June 2000). "Inference of population structure using multilocus genotype data". Genetics. 155 (2): 945–959. ISSN 0016-6731. PMC 1461096. PMID 10835412.
- Verity, Robert; Nichols, Richard A. (August 2016). "Estimating the Number of Subpopulations (K) in Structured Populations". Genetics. 203 (4): 1827–1839. doi:10.1534/genetics.115.180992. ISSN 0016-6731. PMC 4981280. PMID 27317680.
- Manlik, Oliver; Chabanne, Delphine; Daniel, Claire; Bejder, Lars; Allen, Simon J.; Sherwin, William B. (13 November 2018). "Demography and genetics suggest reversal of dolphin source–sink dynamics, with implications for conservation". Marine Mammal Science. 35 (3): 732–759. doi:10.1111/mms.12555.
- Gutenkunst, Ryan N.; Hernandez, Ryan D.; Williamson, Scott H.; Bustamante, Carlos D.; McVean, Gil (23 October 2009). "Inferring the Joint Demographic History of Multiple Populations from Multidimensional SNP Frequency Data". PLOS Genetics. 5 (10): e1000695. doi:10.1371/journal.pgen.1000695. PMC 2760211. PMID 19851460.
- Sniegowski P, Gerrish P, Johnson T, Shaver A; Gerrish; Johnson; Shaver (2000). "The evolution of mutation rates: separating causes from consequences". BioEssays. 22 (12): 1057–1066. doi:10.1002/1521-1878(200012)22:12<1057::AID-BIES3>3.0.CO;2-W. PMID 11084621.CS1 maint: multiple names: authors list (link)
- Lynch, Michael, John S. Conery; Conery (2003). "The origins of genome complexity". Science. 302 (5649): 1401–1404. Bibcode:2003Sci...302.1401L. CiteSeerX 10.1.1.135.974. doi:10.1126/science.1089370. PMID 14631042.CS1 maint: multiple names: authors list (link)
- Rajon, E.; Masel, J. (3 January 2011). "Evolution of molecular error rates and the consequences for evolvability". Proceedings of the National Academy of Sciences. 108 (3): 1082–1087. Bibcode:2011PNAS..108.1082R. doi:10.1073/pnas.1012918108. PMC 3024668. PMID 21199946.
External links
| Wikimedia Commons has media related to Population genetics. |
- Population Genetics Tutorials
- Molecular population genetics
- The ALlele FREquency Database at Yale University
- EHSTRAFD.org - Earth Human STR Allele Frequencies Database
- History of population genetics
- How Selection Changes the Genetic Composition of Population, video of lecture by Stephen C. Stearns (Yale University)
- National Geographic: Atlas of the Human Journey (Haplogroup-based human migration maps)
| Sub-topics | |
|---|---|
| Genetic history by region |
|
| Population genetics by group | |
| |
| Key components | |
|---|---|
| Fields | |
| Archaeogenetics of | |
| Related topics | |
| Lists | |
| |
