Strychnine total synthesis
Strychnine total synthesis in chemistry describes the total synthesis of the complex biomolecule strychnine. The first reported method by the group of Robert Burns Woodward in 1954 is considered a classic in this research field.[2][3][4][5]
At the time it formed the natural conclusion to an elaborate process of molecular structure elucidation that started with the isolation of strychnine from the beans of Strychnos ignatii by Pierre Joseph Pelletier and Joseph Bienaimé Caventou in 1818.[6] Major contributors to the entire effort were Sir Robert Robinson with over 250 publications and Hermann Leuchs with another 125 papers in a time span of 40 years. Robinson was awarded the Nobel Prize in Chemistry in 1947 for his work on alkaloids, strychnine included.
The process of chemical identification was completed with publications in 1946 by Robinson [7][8][9] and later confirmed by Woodward in 1947.[10] X-ray structures establishing the absolute configuration became available between 1947 and 1951 with publications from J. M. Bijvoet [11][12] and J.H. Robertson [13] .[14]
Woodward published a very brief account on the strychnine synthesis in 1954 (just 3 pages) [15] and a lengthy one (42 pages) in 1963.[16]
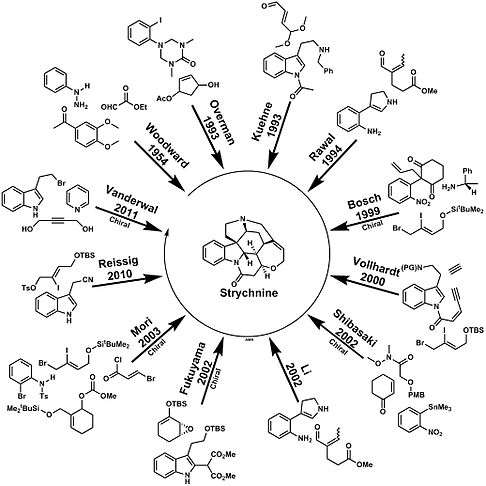
Many more methods exist and reported by the research groups of Magnus,[17] Overman,[18] Kuehne,[19][20] Rawal,[21] Bosch,[22][23] Vollhardt,[24][25] Mori,[26][27] Shibasaki,[28] Li,[29] Fukuyama [30] Vanderwal [31] and MacMillan.[32] Synthetic (+)-strychnine is also known.[33][34] Racemic synthesises were published by Padwa in 2007 [35] and in 2010 by Andrade [36] and by Reissig.[37] In his 1963 publication Woodward quoted Sir Robert Robinson who said [38] for its molecular size it is the most complex substance known.
The molecule
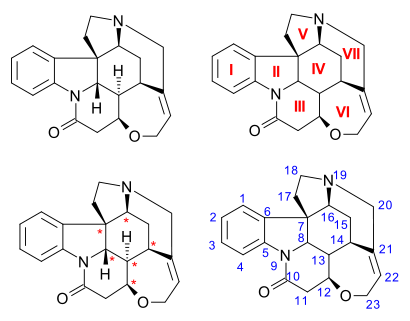
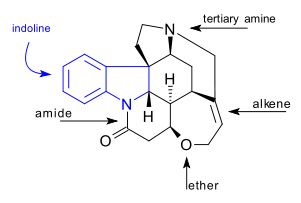
The C21H22N2O2 strychnine molecule contains 7 rings including an indoline system. It has a tertiary amine group, an amide, an alkene and an ether group. The naturally occurring compound is also chiral with 6 asymmetric carbon atoms including one quaternary one.
Woodward synthesis
Ring II, V synthesis
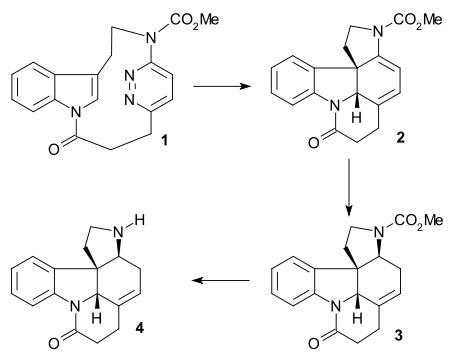
The synthesis of ring II was accomplished with a Fischer indole synthesis using phenylhydrazine 1 and acetophenone derivative acetoveratrone 2 (catalyst polyphosphoric acid) to give the 2-veratrylindole 3. The veratryl group not only blocks the 2-position for further electrophilic substitution but will also become part of the strychnine skeleton. A Mannich reaction with formaldehyde and dimethylamine) produced gramine 4. Alkylation with iodomethane gave an intermediate quaternary ammonium salt which reacted with sodium cyanide in a nucleophilic substitution to nitrile 5 and then in a reduction with lithium aluminium hydride to tryptamine 6. Amine-carbonyl condensation with ethyl glyoxylate give the imine 7. The reaction of this imine with TsCl in pyridine to the ring-closed N-tosyl compound 8 was described by Woodward as a concerted nucleophilic enamine attack and formally a Pictet–Spengler reaction. This compound should form as a diastereomeric pair but only one compound was found although which one was not investigated. Finally the newly formed double bond was reduced by sodium borohydride to indoline 9 with the C8 hydrogen atom approaching from the least hindered side (this proton is removed later on in the sequence and is of no importance).
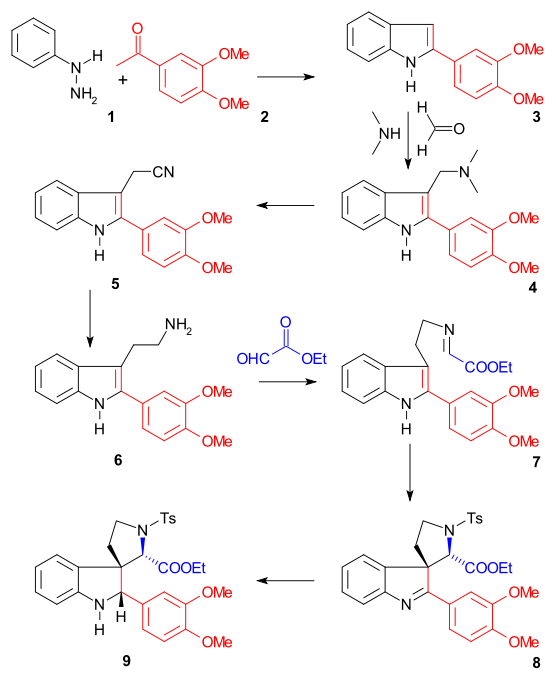
Ring III, IV synthesis
Indoline 9 was acetylated to N-acetyl compound 10 (acetic anhydride, pyridine) and then the veratryl group was then ring-opened with ozone in aquaeous acetic acid to muconic ester 11 (made possible by the two electron-donating methoxide groups). This is an example of bioinspired synthesis already proposed by Woodward in 1948.[39] Cleavage of the acetyl group and ester hydrolysis with HCl in methanol resulted in formation of pyridone ester 12 with additional isomerization of the exocyclic double bond to an endocyclic double bond (destroying one asymmetric center). Subsequent treatment with hydrogen iodide and red phosphorus removed the tosyl group and hydrolysed both remaining ester groups to form diacid 13. Acetylation and esterfication (diazomethane) produced acetyl diester 14 which was then subjected to a Dieckman condensation with sodium methoxide in methanol to enol 15.
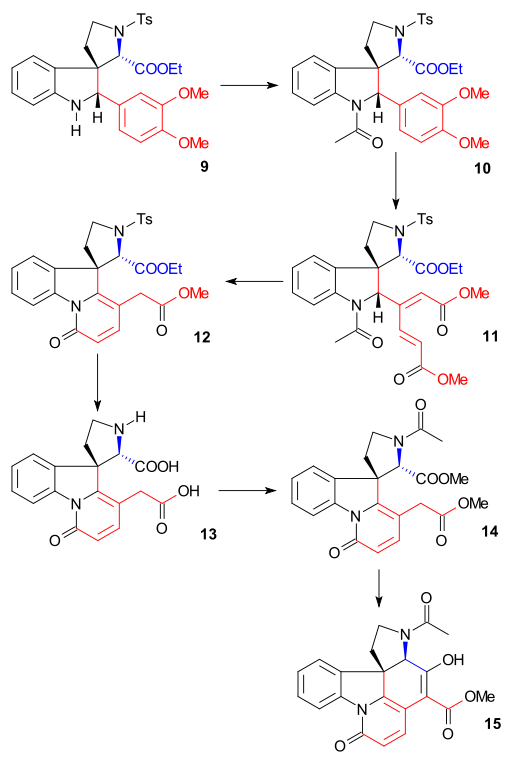
Ring VII synthesis
In order to remove the C15 alcohol group, Enol 15 was converted to tosylate 16 (TsCl, pyridine) and then to mercaptoester 17 (sodium benzylmercaptide) which was then reduced to unsaturated ester 18 by Raney nickel and hydrogen. Further reduction with hydrogen / palladium on carbon afforded the saturated ester 19. Alkaline ester hydrolysis to carboxylic acid 20 was accompanied by epimerization at C14.
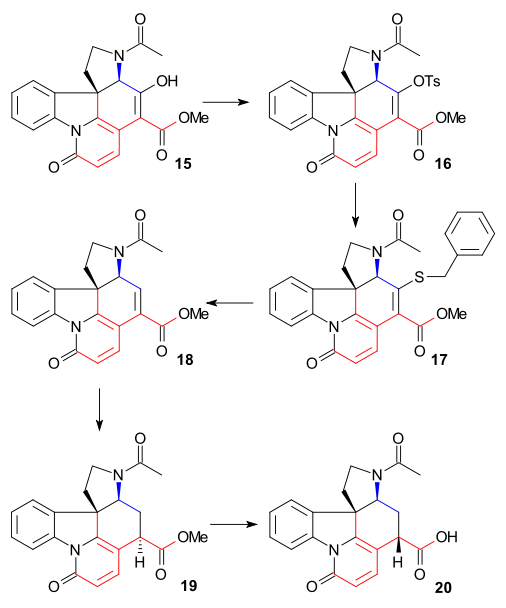
This particular compound was already known from strychnine degradation studies. Until now all intermediates were racemic but chirality was introduced at this particular stage via chiral resolution using quinidine.
The C20 carbon atom was then introduced by acetic anhydride to form enol acetate 21 and the free aminoketone 22 was obtained by hydrolysis with hydrochloric acid. Ring VII in intermediate 23 was closed by selenium dioxide oxidation, a process accompanied by epimerization again at C14.
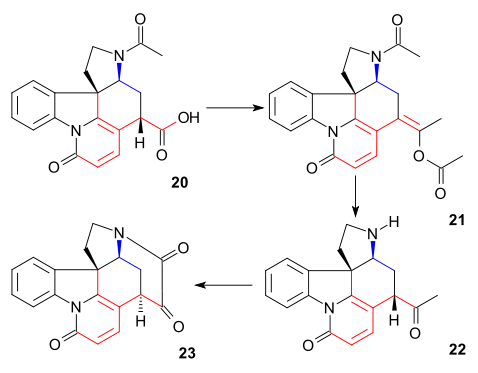
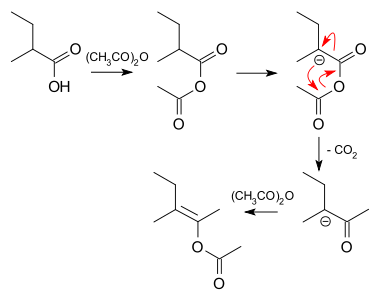
The formation of 21 can be envisioned as a sequence of acylation, deprotonation, rearrangement with loss of carbon dioxide and again acylation:
Ring VI synthesis
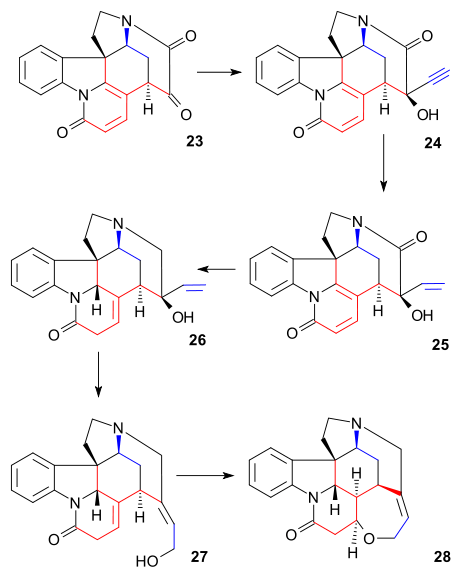
To diketone 23 was added sodium acetylide (bringing in carbon atoms 22 and 23) to give alkyne 24. This compound was reduced to the allyl alcohol 25 using the Lindlar catalyst and lithium aluminium hydride removed the remaining ketone group in 26. An allylic rearrangement to alcohol 27 (isostrychnine) was brought about by hydrogen bromide in acetic acid followed by hydrolysis with sulfuric acid. In the final step to (−)-strychnine 28 treatment of 27 with ethanolic potassium hydroxide caused rearrangement of the C12-13 double bond and ring closure in a conjugate addition by the hydroxyl anion.
Magnus synthesis
In this effort one of strychnine's many degradation products was synthesised first (the relay compound), a compound also available in several steps from another degradation product called the Wieland-Gumlich aldehyde. In the final leg strychnine itself was synthesised from the relay compound.
Overman synthesis
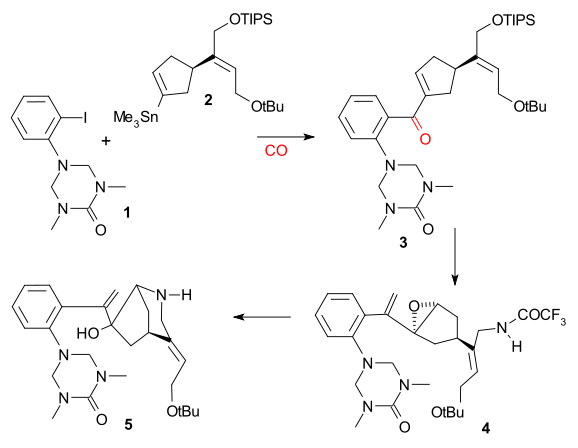
The Overman synthesis (1993) took a chiral cyclopentene compound as starting material obtained by enzymatic hydrolysis of cis-1,4-diacetoxycyclopent-2-ene. This starting material was converted in several steps to trialkylstannane 2 which was then coupled with an aryl iodide 1 in a Stille reaction in presence of carbon monoxide (tris(dibenzylideneacetone)dipalladium(0), triphenylarsine). The internal double in 3 was converted to an epoxide using tert-Butyl hydroperoxide, the carbonyl group was then converted to an alkene in a Wittig reaction using Ph3P=CH2 and the TIPS group was hydrolyzed (TBAF) and replaced by a trifluoroacetamide group (NH2COCF3, NaH) in 4. Cyclization (NaH) took place next, opening the epoxide ring and the trifluoroacetyl group was removed using KOH affording azabicyclooctane 5.
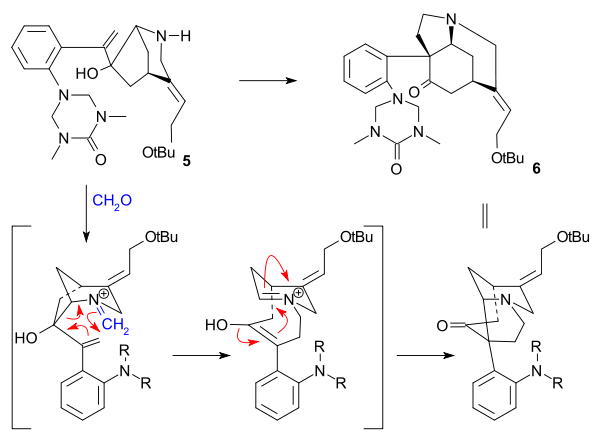
The key step was an aza-Cope Mannich reaction initiated by an amine-carbonyl condensation using formaldehyde and forming 6 in a quantitative yield:
In the final sequence strychnine was obtained through the Wieland-Gumlich aldehyde (10):
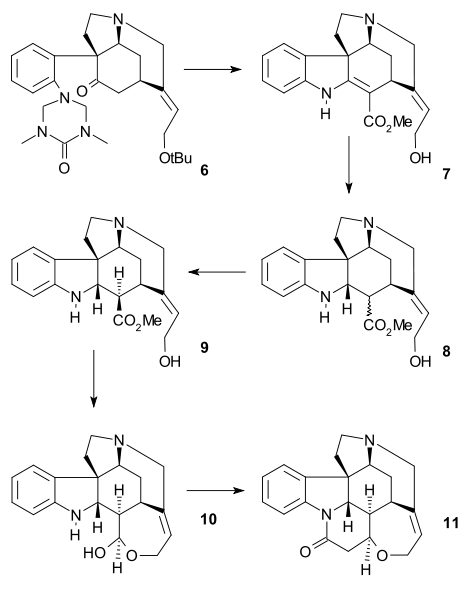
Intermediate 6 was acylated using methyl cyanoformate and two protective groups (tert-butyl and ) were removed using HCl / MeOH in 7. The C8C13 double bond was reduced with zinc (MeOH/H+) to saturated ester 8 (mixture). Epimerization at C13 with sodium methoxide in MeOH produced beta-ester 9 which was reduced with diisobutylaluminium hydride to Wieland-Gumlich aldehyde 10. Conversion of this compound with malonic acid to (−)-strychnine 11 was already known as a procedure.
Kuehne synthesis
The 1993 Keuhne synthesis concerns racemic strychnine. Starting compounds tryptamine 1 and 4,4-dimethoxy acrolein 2 were reacted together with boron trifluoride to acetal 3 as a single diastereomer in an amine-carbonyl condensation / sigmatropic rearrangement sequence.
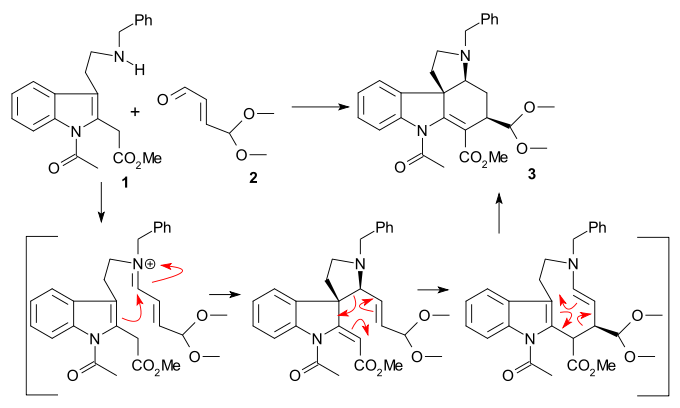
Hydrolysis with perchloric acid afforded aldehyde 4. A Johnson–Corey–Chaykovsky reaction (trimethylsulfonium iodide / n-butyllithium) converted the aldehyde into an epoxide which reacted in situ with the tertiary amine to ammonium salt 5 (contaminated with other cyclization products). Reduction (palladium on carbon/hydrogen) removed the benzyl group to alcohol 6, more reduction (sodium cyanoborohydride) and acylation (acetic anhydride / pyridine) produced 7 as a mixture of epimers (at C17). Ring closure of ring III to 8 was then accomplished with an aldol reaction using lithium bis(trimethylsilyl)amide (using only the epimer with correct configuration). Even more reduction (sodium borohydride) and acylation resulted in epimeric di-acetate 9.
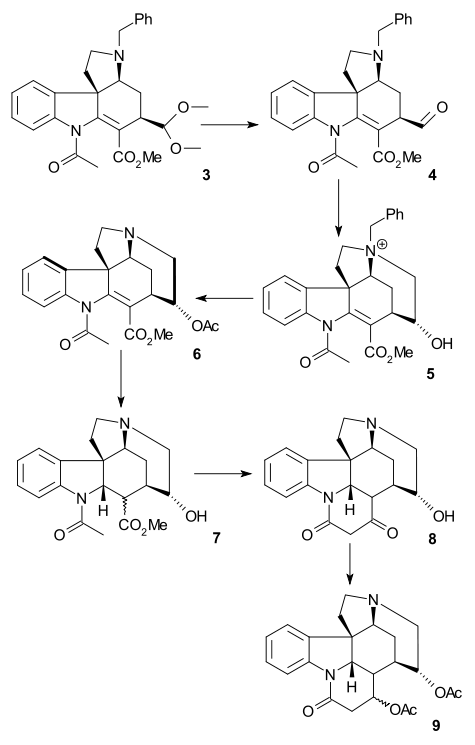
A DBU mediated elimination reaction formed olefinic alcohol 10 and subsequent Swern oxidation have an unstable amino ketone 11. In the final steps a Horner–Wadsworth–Emmons reaction (methyl 2-(diethy1phosphono)acetate) give acrylate ester 12 as a mixture of cis and trans isomers which could be coached into the right (trans) direction by application of light in a photochemical rearrangement, the ester group was reduced (DIBAL / boron trifluoride) to isostrychnine 13 and racemic strychnine 14 was formed by base-catalyzed ring closure as in the Woodward synthesis.
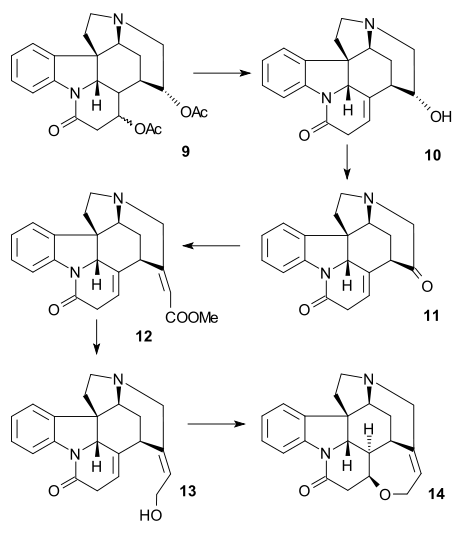
In the 1998 Keuhne synthesis of chiral (−)-strychnine the starting material was derived from chiral tryptophan.
Rawal synthesis
In the Rawal synthesis (1994, racemic) amine 1 and enone 2 were combined in an amine-carbonyl condensation followed by methyl chloroformate quench to triene 3 which was then reacted in a Diels–Alder reaction (benzene 185 °C) to hexene 4. The three ester groups were hydrolyzed using iodotrimethylsilane forming pentacyclic lactam 5 after a methanol quench in a combination of 7 reaction steps (one of them a Dieckmann condensation). The C4 segment 6 was added in an amine alkylation and Heck reaction of 7 formed isostrychnine 8 after TBS deprotection.
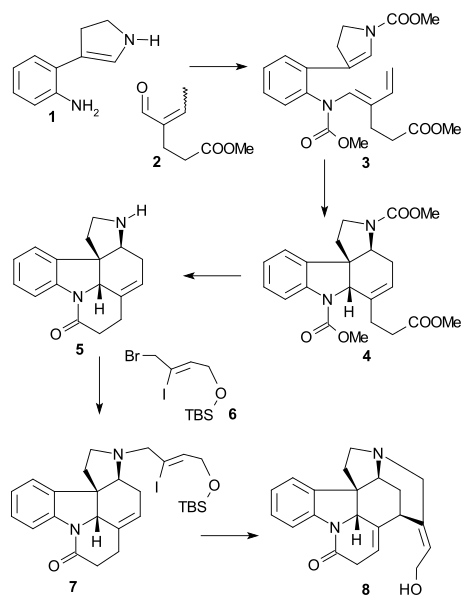
The overall yield (10%) is to date the largest of any of the published methods [40]
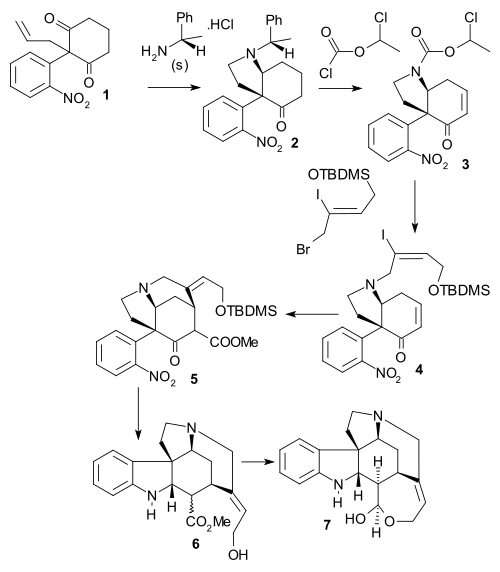
Bosch synthesis
In the Bosch synthesis of (1999, chiral) the olefin group in dione 1 was converted to an aldehyde by ozonolysis and chiral amine 2 was formed in a double reductive amination with (S)-1-phenethylamine. The phenylethyl substituent was removed using ClCO2CHClCH3 and the enone group was introduced in a Grieco elimination using TMSI, HMDS then PhSeCl then ozone and then diisopropylamine forming carbamate 3. The amino group was deprotected by refluxing in methanol and then alkylated using (Z)-BrCH2CICH=CH2OTBDMS, to tertiary amine 4. A reductive Heck reaction took place next followed by methoxycarbonylation (LiHMDS, NCCO2Me) to tricycle 5. Reaction with zinc dust in 10% sulfuric acid removed the TBDMS protective group, reduced the nitro group and brought about a reductive amino-carbonyl cyclization in a single step to tetracyclic 6 (epimeric mixture). In the final step to the Wieland-Gumlich aldehyde 7 reaction with NaH in MeOH afforded the correct epimer was followed by DIBAH reduction of the methyl ester.
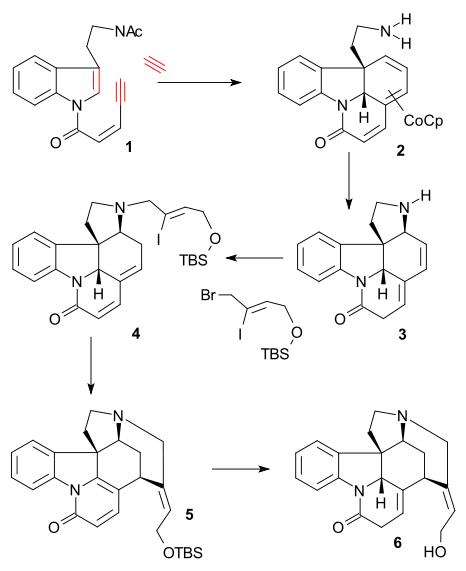
Vollhardt synthesis
The key reaction in the Vollhardt synthesis (2000, racemic) was an alkyne trimerisation of tryptamine derivative 1 with acetylene and organocobalt compound CpCo(C2H4)2 (THF, 0 °C) to tricycle 2 after deprotection of the amine group (KOH, MeOH/H2O reflux). Subsequent reaction with iron nitrate brought about a [1,8]-conjugate addition to tetracycle 3, amine alkylation with (Z)-1-bromo-4-[(tert-butyldimethylsilyl)oxy]-2-iodobut-2-ene (see Rawal synthesis) and lithium carbonate, and isomerization of the diene system (NaOiPr, iPrOH) formed enone 4. A Heck reaction as in the Rawal synthesis (palladium acetate / triphenylphosphine), accompanied by aromatization formed pyridone 5 and lithium aluminium hydride reduction and TBS group deprotection formed isostrychnine 6.
Mori synthesis
The Mori synthesis ((-) chiral, 2003) was the first one containing an asymmetric reaction step. It also features a large number of Pd catalyzed reactions. In it N-tosyl amine 1 reacted with allyl carbonate 2 in an allylic asymmetric substitution using Pd2(dba)3 and asymmetric ligand (S-BINAPO) to chiral secondary amine 3. Desilylation of the TBDMS group next took place by HCl to the hydroxide and then to the nitrile 4 (NaCN) through the bromide (PBr3). Heck reaction (Pd(OAc)2 / Me2PPh) and debromination (Ag2CO3) afforded tricycle 5. LiALH4 Nitrile reduction to the amine and its Boc2O protection to boc amine 6 was then followed by a second allylic oxidation (Pd(OAc)2 / AcOH / benzoquinone / MnO2) to tetracycle 7. Hydroboration-oxidation (9-BBN / H2O2) gave alcohol 8 and subsequent Swern oxidation ketone 9. Reaction with LDA / PhNTf2 gave enol triflate 10 and the triflate group was removed in alkene 11 by reaction with Pd(OAc)2 and PPh3.
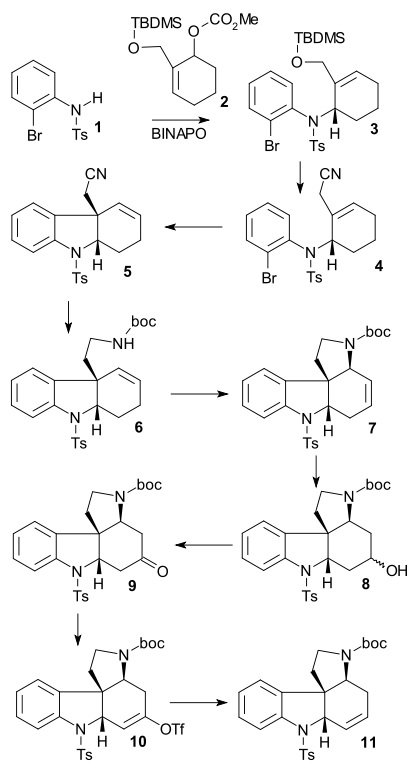
Detosylation of 11 (sodium naphthalenide) and amidation with acid chloride 3-bromoacryloyl chloride gave amide 12 and another Heck reaction gave pentacycle 13. double bond isomerization (sodium / iPrOH), Boc group deprotection (triflic acid) and amine alkylation with (Z)-BrCH2CICH=CH2OTBDMS (see Rawal) gave compound 14 (identical to one of the Vollhardt intermediates). A final heck reaction (15) and TBDMS deprotection formed (−)-isostrychnine 16.
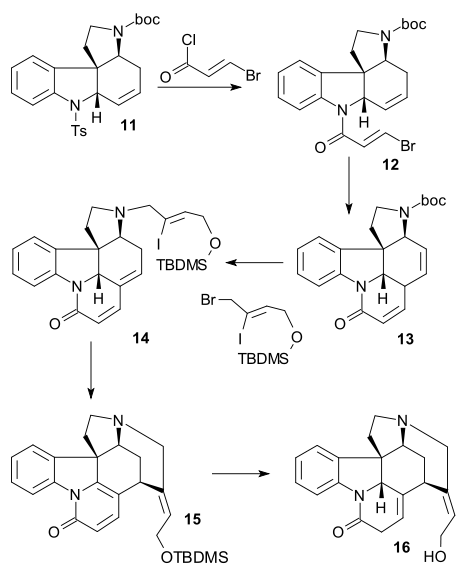
Shibasaki synthesis
The Shibasaki synthesis ((-) chiral, 2002) was a second published method in strychnine total synthesis using an asymmetric reaction step. Cyclohexenone 1 was reacted with dimethyl malonate 2 in an asymmetric Michael reaction using AlLibis(binaphthoxide) to form chiral diester 3. Its ketone group was protected as an acetal (2-ethyl-2-methyl-1,3-dioxolane, TsOH) and a carboxyl group was removed (LiCl, DMSO 140 °C) in monoester 4. A C2 fragment was added as Weinreb amide 5 to form PMB ether 6 using LDA. The ketone was then reduced to the alcohol (NaBH3CN, TiCl4) and then water was eliminated (DCC, CuCl) to form alkene 7. After ester reduction (DIBAL) to the alcohol and its TIPS protection (TIPSOTf, triethylamine), the acetal group was removed (catalytic CSA) in ketone 8. Enone 9 was then formed by Saegusa oxidation. The conversion to alcohol 10 was accomplished via a Mukaiyama aldol addition using formaldehyde, iodonation to 11 (iodine, DMAP) was followed by a Stille coupling (Pd2dba3, Ph3As, CuI) incorporating nitrobenzene unit 12. Alcohol 13 was formed after SEM protection (SEMCl,i-Pr2NEt) and TIPS removal (HF).
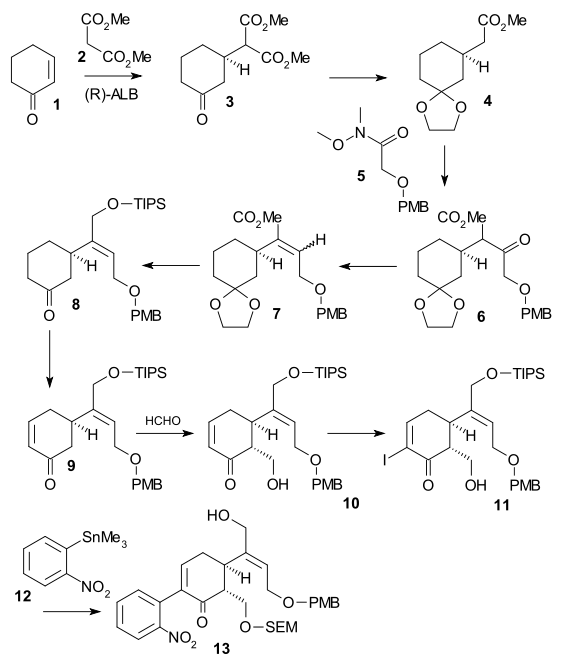
In the second part of the sequence alcohol 13 was converted to a triflate (triflic anhydride, N,N-diisopropylethylamine), then 2,2-bis(ethylthio)ethylamine 14 was added immediately followed by zinc powder, setting of a tandem reaction with nitro group reduction to the amine, 1,4-addition of the thio-amine group and amine-keto condensation to indole 16. Reaction with DMTSF gave thionium attack at C7 forming 17, the imine group was then reduced (NaBH3CN, TiCl4), the new amino group acylated (acetic anhydride, pyridine), both alcohol protecting groups removed (NaOMe / meOH) and the allyl alcohol group protected again (TIPS). This allowed removal of the ethylthio group (NiCl2, NaBH4, EtOH/MeOH) to 18. The alcohol was oxidized to the aldehyde using a Parikh-Doering oxidation and TIPS group removal gave hemiacetal 19 called (+)-diaboline which is acylated Wieland-Gumlich aldehyde.
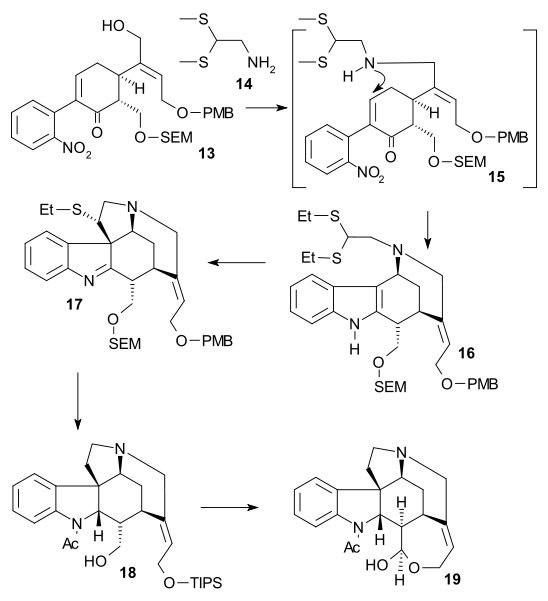
Li synthesis
The synthesis reported by Bodwell/Li (racemic, 2002) was a formal synthesis as it produced a compound already prepared by Rawal (no. 5 in the Rawal synthesis). The key step was an inverse electron demand Diels–Alder reaction of cyclophane 1 by heating in N,N-diethylaniline (dinitrogen is expulsed) followed by reduction of double bond in 2 to 3 by sodium borohydride / triflic acid and removal of the carbamate protecting group (PDC / celite) to 4.
The method is disputed by Reissig (see Reissig synthesis).
Fukuyama synthesis
The Fukuyama synthesis (chiral (-), 2004) started from cyclic amine 1. Chirality was at some point introduced into this starting material by enzymatic resolution of one of the precursors. Acyloin 2 was formed by Rubottom oxidation and hydrolysis. Oxidative cleavage by lead acetate formed aldehyde 3, removal of the nosyl group (thiophenol / cesium carbonate) triggered an amine-carbonyl condensation with iminium ion 4 continuing to react in a transannular cyclization to diester 5 which could be converted to the Wieland-Gumlich aldehyde by known chemistry.
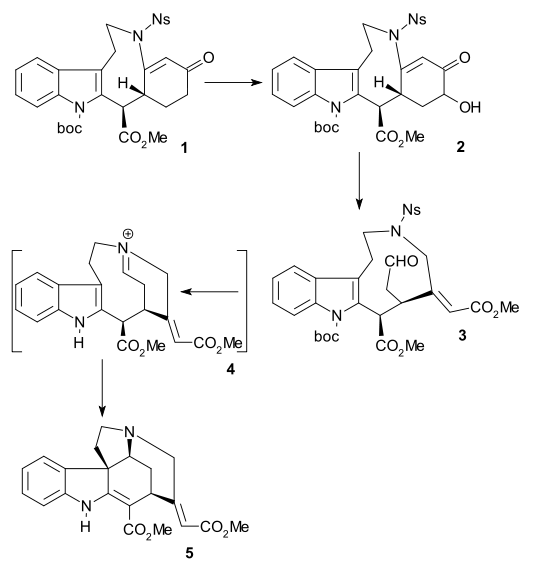
Reissig synthesis
The method reported by Beemelmanns & Reissig (racemic, 2010) is another formal synthesis leading to the Rawal pentacycle (see amine 5 in the Rawal method). In this method indole 1 was converted to tetracycle 2 (together with by-product) in a single cascade reaction using samarium diiodide and HMPA.[41] Raney nickel/ H2 reduction gave amine 3 and a one-pot reaction using methyl chloroformate, DMAP and TEA then MsCl, DMAP and TEA and then DBU gave Rawal precursor 4 with key hydrogen atoms in the desired anti configuration.
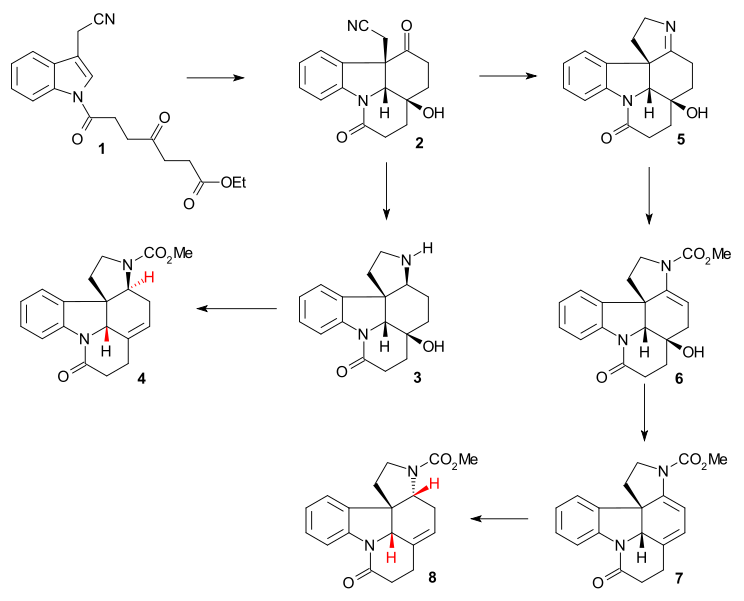
In an aborted route intermediate 2 was first reduced to imine 5 then converted to carbamate 6, then dehydrated to diene 7 (Burgess reagent) and finally reduced to 8 (sodium cyanoborohydride). The hydrogen atoms in 8 are in an undesired cis-relationship which contradicts the results obtained in 2002 by Bodwell/Li for the same reaction.
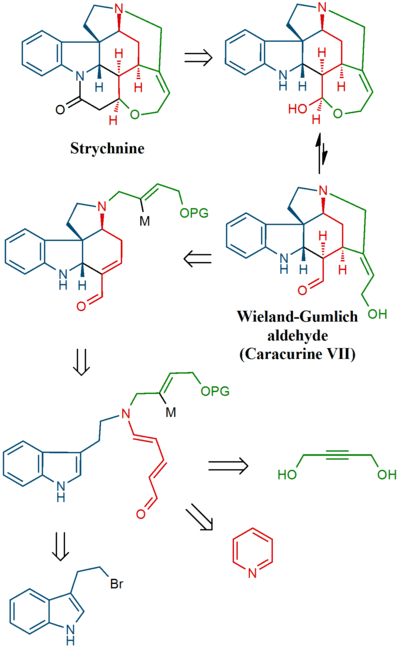
Vanderwal synthesis
In 2011, the Vanderwal group reported a concise, longest linear sequence of 6 steps, total synthesis of strychnine.[42] It featured a Zincke reaction followed by an anionic bicyclization reaction and a tandem Brook rearrangement / conjugate addition.
External links
References
- X-ray; Messerschmidt, M.; Scheins, S.; Luger, P. (2005). "Charge density of (−)-strychnine from 100 to 15 K, a comparison of four data sets". Acta Crystallogr B. 61 (1): 115–121. doi:10.1107/S0108768104032781.
- Nicolaou, K. C.; Sorensen, E. J. (1996). Classics in Total Synthesis: Targets, Strategies, Methods. Wiley. ISBN 978-3-527-29231-8.
- K. C. Nicolaou, Dionisios Vourloumis, Nicolas Winssinger, Phil S. Baran The Art and Science of Total Synthesis at the Dawn of the Twenty-First Century Angewandte Chemie International Edition 2000; Volume 39, Issue 1, Pages: 44-122
- Bonjoch, Josep; Sole, Daniel (2000). "Synthesis of Strychnine". Chem. Rev. 100 (9): 3455–3482. doi:10.1021/cr9902547. PMID 11777429.
- Proudfoot, John R. (2013). "Reaction Schemes Visualized in Network Form: The Syntheses of Strychnine as an Example". Journal of Chemical Information and Modeling. 53 (5): 1035–1042. doi:10.1021/ci300556b.
- Pelletier; Caventou (1818). "Note sur un nouvel alkalai (Note on a new alkali)". Annales de Chimie et de Physique. 8: 323–324. See also: Pelletier; Caventou (1819). "Mémoire sur un nouvel alcali vegetal (la strychnine) trouvé dans la feve de Saint-Ignace, la noix vomique, etc. (Memoir on a new vegetable alkali (strychnine) found in the St. Ignatius bean, the nux vomica, etc)". Annales de Chimie et de Physique. 10: 142–176.
- Robinson, R. (1946). "The constitution of strychnine". Experientia. 2 (1): 1946. doi:10.1007/BF02154708.
- Briggs, L. H.; Openshaw, H. T.; Robinson, Robert (1946). "Strychnine and brucine. Part XLII. Constitution of the neo-series of bases and their oxidation products". J. Chem. Soc. 1946: 903. doi:10.1039/JR9460000903.
- Openshaw, H. T.; Robinson, R. (1946). "Constitution of Strychnine and the Biogenetic Relationship of Strychnine and Quinine". Nature. 157 (3988): 438. Bibcode:1946Natur.157..438O. doi:10.1038/157438a0.
- Woodward, R. B.; Brehm, Warren J.; Nelson, A. L. (1947). "THE STRUCTURE OF STRYCHNINE". J. Am. Chem. Soc. 69 (9): 2250. doi:10.1021/ja01201a526.
- Bijvoet , Schoone and Bokhoven , Kon. Ned. Akad. Wet., 50, No 8, 51, No. 8, 52, No. 2 (1947–49)
- Bokhoven, C.; Schoone, J. C.; Bijvoet, J. M. (1951). "The Fourier synthesis of the crystal structure of strychnine sulphate pentahydrate" (PDF). Acta Crystallogr. 4 (3): 275–280. doi:10.1107/S0365110X51000891.
- Robertson, J. H.; Beevers, C. A. (1950). "Crystal Structure of Strychnine Hydrobromide". Nature. 165 (4200): 690–691. Bibcode:1950Natur.165..690R. doi:10.1038/165690a0.
- Robertson, J. H.; Beevers, C. A. (1951). "The crystal structure of strychnine hydrogen bromide". Acta Crystallogr. 4 (3): 270–275. doi:10.1107/S0365110X5100088X.
- Woodward, R. B.; Cava, Michael P.; Ollis, W. D.; Hunger, A.; Daeniker, H. U.; Schenker, K. (1954). "THE TOTAL SYNTHESIS OF STRYCHNINE". J. Am. Chem. Soc. 76 (18): 4749–4751. doi:10.1021/ja01647a088.
- Woodward, R. B.; Cava, M. P.; Ollis, W. D.; Hunger, A.; Daeniker, H. U.; Schenker, K. (1963). "The total synthesis of strychnine". Tetrahedron. 19 (2): 247–288. doi:10.1016/s0040-4020(01)98529-1.
- Magnus, Philip; Giles, Melvyn; Bonnert, Roger; Kim, Chung S.; McQuire, Leslie; Merritt, Andrew; Vicker, Nigel (1992). "Synthesis of strychnine via the Wieland-Gumlich aldehyde". J. Am. Chem. Soc. 114 (11): 4403–4405. doi:10.1021/ja00037a058.
- Knight, Steven D.; Overman, Larry E.; Pairaudeau, Garry (1993). "Synthesis applications of cationic aza-Cope rearrangements. 26. Enantioselective total synthesis of (−)-strychnine". J. Am. Chem. Soc. 115 (20): 9293–9294. doi:10.1021/ja00073a057.
- Kuehne, Martin E.; Xu, Feng (1993). "Total synthesis of strychnan and aspidospermatan alkaloids. 3. The total synthesis of (+-)-strychnine". J. Org. Chem. 58 (26): 7490–7497. doi:10.1021/jo00078a030.
- Kuehne, Martin E.; Xu, Feng (1998). "Syntheses of Strychnan- and Aspidospermatan-Type Alkaloids. 10. An Enantioselective Synthesis of (−)-Strychnine through the Wieland−Gumlich Aldehyde". J. Org. Chem. 63 (25): 9427–9433. doi:10.1021/jo9813989.
- Rawal, Viresh H.; Iwasa, Seiji (1994). "A Short, Stereocontrolled Synthesis of Strychnine". J. Org. Chem. 59 (10): 2685–2686. doi:10.1021/jo00089a008.
- Total Synthesis of (−)-Strychnine via the Wieland-Gumlich Aldehyde Angewandte Chemie International Edition Volume 38, Issue 3, 1999, Pages: 395-397 Daniel Solé, Josep Bonjoch, Silvina García-Rubio, Emma Peidró, Joan Bosch
- Solé, Daniel; Bonjoch, Josep; García-Rubio, Silvina; Peidró, Emma; Bosch, Joan (2000). "Enantioselective Total Synthesis of Wieland-Gumlich Aldehyde and (−)-Strychnine". Chemistry: A European Journal. 6 (4): 655–665. doi:10.1002/(SICI)1521-3765(20000218)6:4<655::AID-CHEM655>3.0.CO;2-6.
- Eichberg, Michael J.; Dorta, Rosa L.; Lamottke, Kai; Vollhardt, K. Peter C. (2000). "The Formal Total Synthesis of (±)-Strychnine via a Cobalt-Mediated [2 + 2 + 2]Cycloaddition". Org. Lett. 2 (16): 2479–2481. doi:10.1021/ol006131m.
- Eichberg, Michael J.; Dorta, Rosa L.; Grotjahn, Douglas B.; Lamottke, Kai; Schmidt, Martin; Vollhardt, K. Peter C. (2001). "Approaches to the Synthesis of (±)-Strychnine via the Cobalt-Mediated [2 + 2 + 2] Cycloaddition: Rapid Assembly of a Classic Framework". J. Am. Chem. Soc. 123 (38): 9324–9337. doi:10.1021/ja016333t.
- Nakanishi, Masato; Mori, Miwako (2002). "Total Synthesis of (−)-Strychnine". Angewandte Chemie International Edition. 41 (11): 1934–1936. doi:10.1002/1521-3773(20020603)41:11<1934::AID-ANIE1934>3.0.CO;2-F.
- Mori, Miwako; Nakanishi, Masato; Kajishima, Daisuke; Sato, Yoshihiro (2003). "A Novel and General Synthetic Pathway to Strychnos Indole Alkaloids: Total Syntheses of (−)-Tubifoline, (−)-Dehydrotubifoline, and (−)-Strychnine Using Palladium-Catalyzed Asymmetric Allylic Substitution". J. Am. Chem. Soc. 125 (32): 9801–9807. doi:10.1021/ja029382u.
- Ohshima, Takashi; Xu, Youjun; Takita, Ryo; Shimizu, Satoshi; Zhong, Dafang; Shibasaki, Masakatsu (2002). "Enantioselective Total Synthesis of (−)-Strychnine Using the Catalytic Asymmetric Michael Reaction and Tandem Cyclization". J. Am. Chem. Soc. 124 (49): 14546–14547. doi:10.1021/ja028457r.
- Bodwell, Graham J.; Li, Jiang (2002). "A Concise Formal Total Synthesis of (±)-Strychnine by Using a Transannular Inverse-Electron-Demand Diels–Alder Reaction of a [3](1,3)Indolo[3](3,6)pyridazinophane". Angewandte Chemie International Edition. 41 (17): 3261–3262. doi:10.1002/1521-3773(20020902)41:17<3261::AID-ANIE3261>3.0.CO;2-K.
- Kaburagi, Y; Tokuyama, H; Fukuyama, T (2004). "Total synthesis of (−)-strychnine". J. Am. Chem. Soc. 126 (33): 10246–10247. doi:10.1021/ja046407b. PMID 15315428.
- Martin, David B. C.; Vanderwal, Christopher D. (2011). "A synthesis of strychnine by a longest linear sequence of six steps". Chemical Science. 2 (4): 649. doi:10.1039/C1SC00009H.
- Jones, Spencer B.; Simmons, Bryon; Mastracchio, Anthony; MacMillan, David W. C. (2011). "Collective synthesis of natural products by means of organocascade catalysis". Nature. 475 (7355): 183–188. doi:10.1038/nature10232. PMC 3439143. PMID 21753848.
- Knight, Steven D.; Overman, Larry E.; Pairaudeau, Garry (1995). "Asymmetric Total Syntheses of (−)- and (+)-Strychnine and the Wieland-Gumlich Aldehyde". J. Am. Chem. Soc. 117 (21): 5776–5788. doi:10.1021/ja00126a017.
- Not counted:an unpublished method by Gilbert Stork, Lecture at the Ischia School of Organic Chemistry, Ischia Porb, Italy, September 211992.
- Zhang, Hongjun; Boonsombat, Jutatip; Padwa, Albert (2007). "Total Synthesis of (±)-Strychnine via a [4 + 2]-Cycloaddition/Rearrangement Cascade". Org. Lett. 9 (2): 279–282. doi:10.1021/ol062728b. PMC 2587098. PMID 17217284.
- Sirasani, Gopal; Paul, Tapas; William Dougherty, Jr.; Kassel, Scott; Andrade, Rodrigo B. (2010). "Concise Total Syntheses of (±)-Strychnine and (±)-Akuammicine". The Journal of Organic Chemistry. 75 (10): 3529–3532. doi:10.1021/jo100516g. PMID 20408591.
- Beemelmanns, C.; Reissig, H.-U. (2010). "A Short Formal Total Synthesis of Strychnine with a Samarium Diiodide Induced Cascade Reaction as the Key Step". Angewandte Chemie International Edition. 49 (43): 8021–8025. doi:10.1002/anie.201003320. PMID 20848626.
- R. Robinson "Molecular structure of Strychnine, Brucine and Vomicine Prog. Org. Chem., 1952; 1 ,2
- Woodward, R. B. (1948). "Biogenesis of the Strychnos Alkaloids". Nature. 162 (4108): 155–156. Bibcode:1948Natur.162..155W. doi:10.1038/162155a0.
- Cannon, J. S.; Overman, L. E. (2012). "Is There No End to the Total Syntheses of Strychnine? Lessons Learned in Strategy and Tactics in Total Synthesis". Angew. Chem. Int. Ed. 51 (18): 4288–4311. doi:10.1002/anie.201107385. PMC 3804246. PMID 22431197.
- Szostak, M.; Procter, D. J. (2011). "Concise Syntheses of Strychnine and Englerin A: the Power of Reductive Cyclizations Triggered by Samarium Iodide". Angewandte Chemie International Edition. 50 (34): 7737–7739. doi:10.1002/anie.201103128. PMID 21780264.
- Martin, David B. C.; Vanderwal, Christopher D. (2011). "A synthesis of strychnine by a longest linear sequence of six steps". Chemical Science. 2 (4): 649. doi:10.1039/C1SC00009H.