Rubottom oxidation
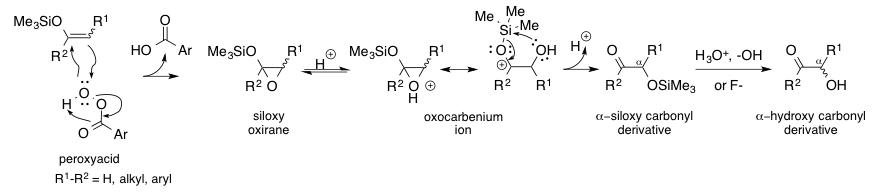
The Rubottom oxidation is a useful, high-yielding chemical reaction between silyl enol ethers and peroxyacids to give the corresponding α-hydroxy carbonyl product.[1][2][3][4][5] The mechanism of the reaction was proposed in its original disclosure by A.G. Brook[6][7] with further evidence later supplied by George M. Rubottom.[8] After a Prilezhaev-type oxidation of the silyl enol ether with the peroxyacid to form the siloxy oxirane intermediate, acid-catalyzed ring-opening yields an oxocarbenium ion.[1][4] This intermediate then participates in a 1,4-silyl migration (Brook rearrangement) to give an α-siloxy carbonyl derivative that can be readily converted to the α-hydroxy carbonyl compound in the presence of acid, base, or a fluoride source.[1][9][10]
| Rubottom oxidation | |
|---|---|
| Named after | George M. Rubottom |
| Reaction type | Organic redox reaction |
| Identifiers | |
| Organic Chemistry Portal | rubottom-oxidation |
Reaction mechanism
History
In 1974, three independent groups reported on the reaction now known as the Rubottom oxidation:[1] A.G Brook,[6] A. Hassner,[11] and G.M. Rubottom.[12] Considerable precedent for the reaction already existed.[3] For instance, it was known as early as the 1930s that highly enolizable β-dicarbonyl compounds would react with peroxyacids, although it was not until the 1950s and 60s α-hydroxy β-dicarbonyl compounds were in fact the product.[13][14]
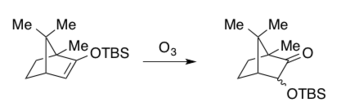
Considerable work by A.G Brook, during the 1950s on the mechanisms of organosilicon migrations, which are now known as Brook Rearrangements.[15][16] In 1974, C.H. Heathcock described the ozonolysis of silyl enol ethers to give a carboxylic acid product via oxidative cleavage where silyl migrations were observed as side reactions and exclusively in the case of a bicyclic system.[17]
General features
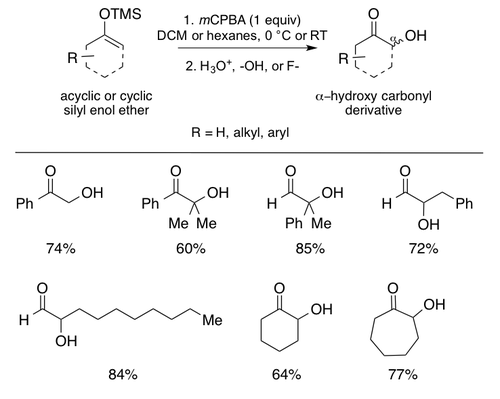
The original implementations of the Rubottom oxidation featured the peroxyacid meta-chloroperoxybenzoic acid (mCPBA) as the oxidant in dichloromethane (DCM), in the case of Hassner and Brook, and hexanes for Rubottom.[6][11][12] While the reaction has been tweaked and modified since 1974, mCPBA is still commonly used as the oxidant with slightly more variation in the solvent choice.[1][4] DCM remains the most common solvent followed by various hydrocarbon solvents including pentane and toluene.[1][4] Notably, the reaction proceeds at relatively low temperatures and heating beyond room temperature is not necessary.[1][4] Low temperatures allow the standard Rubottom oxidation conditions to be amenable with a variety of sensitive functionalities making it ideal for complex molecule synthesis (See synthetic examples below). Silyl enol ether substrates can be prepared regioselectively from ketones or aldehydes by employing thermodynamic or kinetic control to the enolization prior to trapping with the desired organosilicon source (usually a chloride or triflate e.g. TBSCl or TBSOTf).[18] As illustrated by the synthetic examples below, silyl enol ethers can be isolated prior to exposure to the reaction conditions, or the crude material can be immediately subjected to oxidation without isolation. Both acyclic and cyclic silyl enol ether derivatives can be prepared in this way and subsequently be used as substrates in the Rubottom oxidation.[1] Below are some representative Rubottom oxidation products synthesized in the seminal papers.[6][11][12]
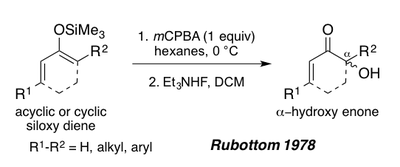
In 1978, Rubottom showed that siloxy 1,3 dienes, derived from acyclic or cyclic enones could also serve as substrates for the Rubottom oxidation to forge α-hydroxy enones after treatment with triethyl ammonium fluoride.[1][19] These substrates give a single regioisomer under the reaction conditions due to the electron-rich nature of the silyl enol pi-bond (See synthesis of Periplanone B below).[1]
Modifications and Improvements
The Rubottom oxidation has remained largely unchanged since its initial disclosure, but one of the major drawbacks of standard conditions is the acidic environment, which can lead to unwanted side reactions and degradation. A simple sodium bicarbonate buffer system is commonly employed to alleviate this issue, which is especially problematic in bicyclic and other complex molecule syntheses (see synthetic examples).[1][20] The introduction of chiral oxidants has also allowed for the synthesis of enantiopure α-hydroxy carbonyl derivatives from their corresponding silyl enol ethers.[1] The first example of an enantioselective Rubottom oxidation was published by F.A. Davis[21] in 1987 and showcased the Davis chiral oxaziridine methodology to give good yields but modest enantiomeric excesses. In 1992, K.B. Sharpless showed that the asymmetric dihydroxylation conditions developed in his group could be harnessed to give either (R)- or (S)- α-hydroxy ketones from the corresponding silyl enol ethers depending on which Chinchona alkaloid-derived chiral ligands were employed.[22] The groups of Y. Shi[23] and W. Adam[24] published another enantioselective variant of the Rubottom oxidation in 1998 using the Shi chiral ketone in the presence of oxone in a buffered system to furnish α-hydroxy ketones in high yield and high enantiomeric excess. The Adam group also published another paper in 1998 utilizing manganese(III)-(Salen)complexes in the presence of NaOCl (bleach) as the oxidant and 4-phenylpyridine N-oxide as an additive in a phosphate buffered system.[25] This methodology also gave high yields and enentioselectivities for silyl enol ethers as well as silyl ketene acetals derived from esters.
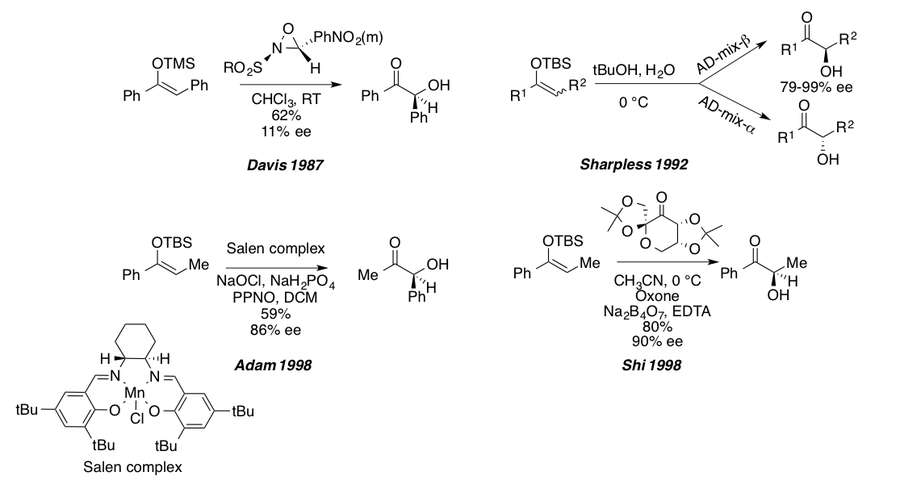
Along with chiral oxidants, variants of mCPBA have been examined.[1] Stankovic and Espenson published a variation of the Rubottom oxidation where methyltrioxorhenium is used as a catalytic oxidant in the presence of stoichiometric hydrogen peroxide.[1][26] This methodology gives acyclic and cyclic α-hydroxy ketones in high yield with a cheap, commercially available oxidant. An inherent problem with mCPBA is its inability to oxidize silyl ketene acetals. In order to synthesize α-hydroxy esters, different oxidants are needed such as NaOCl (see above), lead(IV) acetate, or a hypofluorous acid-acetonitrile (HOF-ACN) complex.[1][27] The Rubottom group found that lead(IV) acetate in DCM or benzene gave good yields of acyclic and cyclic α-hydroxy esters after treatment of the crude reaction mixture with triethylammonium fluoride.[27] Later, the highly electrophilic HOF-ACN complex was used by S. Rozen to oxidize a variety of electron rich silyl enol ethers, silyl ketene acetals, and bis(silyl acetals), derived from carboxylic acids, in good yields at or below room temperature.[1][28]
Applications in synthesis
The following examples represent only a small portion of syntheses that highlight the use of the Rubottom oxidation to install an important α-hydroxy functionality. Some of the major features of the following syntheses include the use of buffered conditions to protect sensitive substrates and the diastereoselective installation of the α-hydroxy group due to substrate controlled facial bias. For more examples see refs[1][3][4]
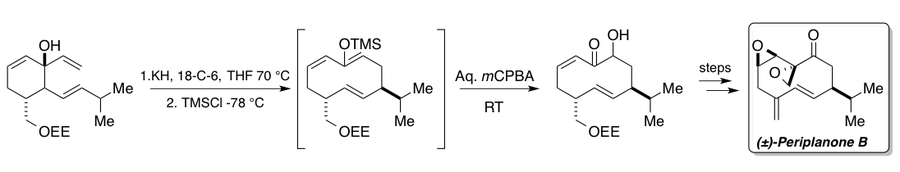
The Rubottom oxidation was used in the synthesis of periplanone B, a sex pheromone excreted by the female American cockroach.[29][30] The synthesis employed an anionic oxy-Cope rearrangement coupled to a Rubottom oxidation. After heating in the presence of potassium hydride (KH) and 18-crown-6 (18-C-6) to effect the anionic oxy-Cope, the enolate intermediate was trapped with trimethylsilyl chloride (TMSCl). The silyl enol ether intermediate could then be treated with mCPBA under Rubottom oxidation conditions to give the desired α-hydroxy carbonyl compound that could then be carried on to (±)-periplanone B and its diastereomers to prove its structure.
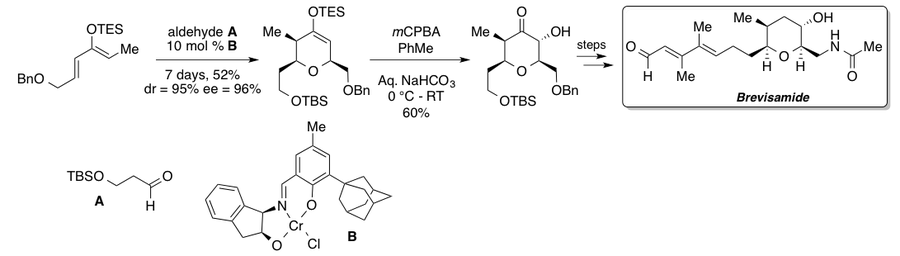
Brevisamide, a proposed biosynthetic precursor for a polyether marine toxin, was synthesized by Ghosh and Li, one step of which is a Rubottom oxidation of the cyclic silyl enol ether under buffered conditions.[31] Chiral chromium catalyst B was developed the Jacobsen group and confers high levels of enantio- and diastereoselectivity.[32] The stereocenters conveniently set in the Diels-Alder reaction direct the oxidation to the less hindered face, giving a single diastereomer, which could then be carried on in 14 more steps to Brevisamide.
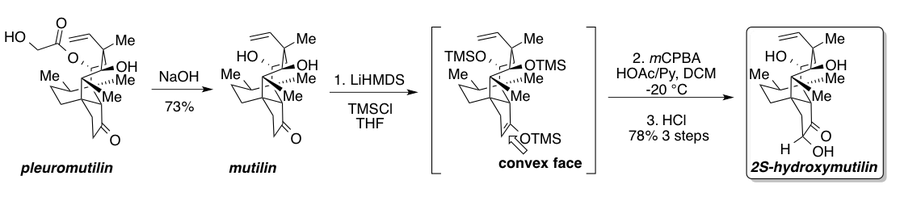
Wang and coworkers developed a robust, kilogram-scale synthesis of the potent derivative 2S-hydroxymutilin from pleuromutilin, an antibiotic produced by various species of basidiomycetes.[33] Basic hydrolysis to remove the hydroxyl ester moiety of pleuromutilin yielded mutilin. Subsequent treatment with lithium hexamethyldisilazide (LiHMDS) and TMSCl gave the TMS-protected silyl enol ether, which was immediately subjected to an acetic acid- (HOAc) pyridine- (Py) buffered Rubottom oxidation before acidic hydrolysis to afford 2S-hydroxymutilin. This highly optimized sequence features two important aspects. First, the authors originally generated the silyl enol ether using triethylamine, which gave a mixture of the desired kinetic product, (shown below) the undesired thermodynamic product, and hydrolysis back to mutilin. The authors blamed the formation of the acidic triethylammonium (pKa = 10.6) byproduct for the undesired side products and remedied this by using the LiHMDS to exclusively form the desired kinetic product with no acid-catalyzed side reactions due to the significantly lower acidity of the protonated product (pKa = 26).[34] Second, while oxidation occurred from the desired convex face of the silyl enol ether, the authors saw a significant number of overoxidation products that they attributed to the stability of the oxocarbenium ion intermediate under sodium bicarbonate buffered conditions. They hypothesized that the increased lifetime of the intermediate species would allow for over oxidation to occur. After a significant amount of optimization, it was found that an HOAc/Py buffer trapped the oxocarbenium intermediate and prevented overoxidation to exclusively give 2S-hydroxymutilin after hydrolysis of the silyl protecting groups.
Ovalicin, fumagillin, and their derivatives exhibit strong anti-angiogenesis properties and have seen numerous total syntheses since their isolation.[35] Corey and Dittami reported the first total synthesis of racemic ovalicin in 1985[36] followed by two asymmetric syntheses reported in 1994 by Samadi[37][38] and Corey[39] which featured a chiral pool strategy from L-quebrachitol and an asymmetric dihydroxylation, respectively. In 2010, Yadav and coworkers reported a route that intercepted the Samadi route from the chiral pool starting material D-ribose.[40] A standard Rubottom oxidation gives a single stereoisomer due to substrate control and represents the key stereogenic step in the route to the Samadi ketone. Once synthesized, the Samadi ketone could be elaborated to (−)-ovalicin through known steps.
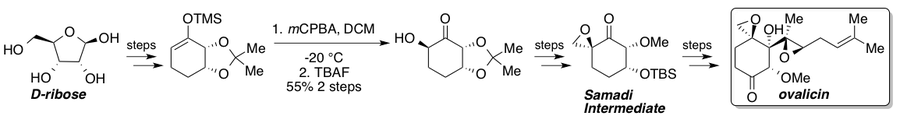
Velutinol A[41] was first synthesized by Isaka and coworkers.[42] The authors show that the high regioselectivity of this reaction is directed by the hydroxyl group syn to the ring-fusion proton. Reactions where the stereochemistry of the hydroxyl group is inverted saw lower regioselectivity, and removal of the hydroxyl group gave the exclusive formation of the other regioisomer. It is likely that the close proximity of the hydroxyl group in the syn isomer acidifies the ring-fusion proton through hydrogen-bonding interactions, thus facilitating regioselective deprotonation by triethylamine. The silyl enol ether was then treated with excess mCPBA to facilitate a “double” Rubottom oxidation to give the exo product with both hydroxyl groups on the outside of the fused ring system. This dihydroxy product was then transformed into Velutinol A in three additional steps.
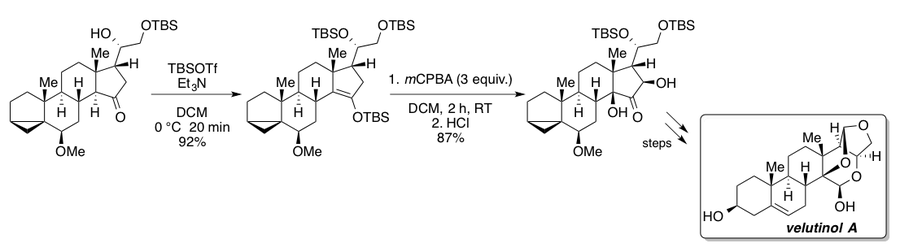
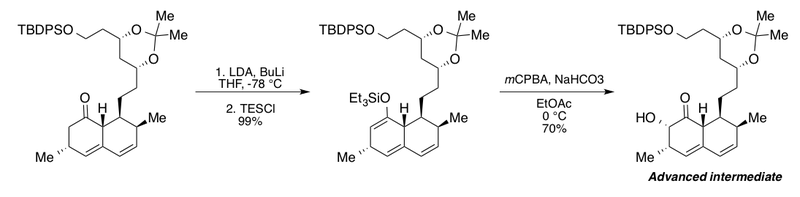
The Clive group utilized the Rubottom oxidation in the synthesis of an advanced intermediate for their degradation studies of the cholesterol-lowering fungal metabolite mevinolin.[2][43] This interesting sequence features the addition of excess n-butyllithium (BuLi) in the presence of lithium diisopropylamide (LDA) for full conversion of the bicyclic ketone derivative to the corresponding silyl enol ether. Without BuLi the authors report a maximum yield of only 72%. Subsequent buffered Rubottom oxidation conditions with sodium bicarbonate in ethyl acetate afforded the α-hydroxy ketone as a single diastereomer.
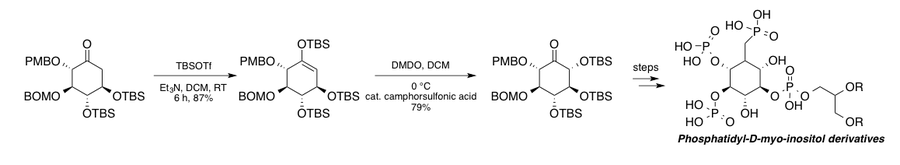
The Falk group synthesized various derivatives of phosphatidyl-D-myo-inositol to aid in the study of the various phosphatidylinositol 3-kinase (PI3K) cell signaling pathways.[2][44] Their route to the collection of substrate analogs exploits a substrate-controlled stereoselective Rubottom oxidation using dimethyl dioxirane(DMDO) as the oxidant and catalytic camphorsulfonic acid (CSA) to aid in hydrolysis. For protecting groups see ref[10]
Problems and shortcomings
While the Rubottom oxidation generally gives good yields and is highly scalable (see 2S-hydroxymutilin synthesis), there are still some problems with the reaction. As mentioned above, the acidic reaction conditions are not tolerated by many complex substrates, but this can be abrogated with the use of buffer systems.[1] Poor atom economy is also a major issue with the reaction because it requires stoichiometric oxidant, which generates large amounts of waste.[3] Peroxides can also be dangerous to work with. mCPBA is known to detonate from shock or sparks.[45]
α-Hydroxylation of related compounds
Although silyl enol ethers of aldehydes and ketones are the traditional substrates for the Rubottom oxidation, as mentioned above, silyl ketene acetals and bis (silyl acetals) can be oxidized to their α-hydroxy ester or carboxylic acid derivatives using lead(IV) acetate or hypofluorous acid-acetonitrile (HOF–ACN).[27] However, these α-hydroxylations do not proceed via silyl enol ether intermediates and are therefore not technically Rubottom oxidations. Various oxidants can be used to oxidize many of these carbonyl derivatives after they are converted to their respective enolate or related anion. Some common oxidants are peroxy acids, molecular oxygen, and hypervalent iodine reagents.[5]
References
- Kürti, pp. 388–389.
- Myers, A.G. Chemistry 215: Oxidation Archived 2011-03-12 at the Wayback Machine. chem.harvard.edu
- Christoffers, J.; Baro, A.; Werner, T. (2004). "α-Hydroxylation Of β-Dicarbonyl Compounds". Adv. Synth. Catal. 346 (23): 143–151. doi:10.1002/adsc.200303140.
- Li, pp. 478–479.
- Chen, B. C.; Zhou, P.; Davis, F. A.; Ciganek, E. (2003) “α-Hydroxylation of Enolates and Silyl Enol Ethers." in Organic Reactions; Ed. Overman, L.E. Wiley, Chapter 1, pp. 1–355, doi:10.1002/0471264180.or062.01.
- Brook, A. G.; Macrae, D. M. (1974). "1, 4-Silyl Rearrangements of Siloxyalkenes to Siloxyketones During Peroxidation". J. Organomet. Chem. 77 (2): C19–C21. doi:10.1016/S0022-328X(00)81332-7.
- Brook, A. G. (1974). "Molecular Rearrangements of Organosilicon Compounds". Acc. Chem. Res. 7 (3): 77–84. doi:10.1021/ar50075a003.
- Rubottom, G. M.; Gruber, J. M.; Boeckman, R. K., Jr; Ramaiah, M.; Medwid, J. B. (1978). "Clarification of the Mechanism of Rearrangement of Enol Silyl Ether Epoxides". Tetrahedron Lett. 19 (47): 4603–4606. doi:10.1016/S0040-4039(01)85682-3.CS1 maint: multiple names: authors list (link)
- Myers, A.G. Chemistry 215: Protective Groups-Silicon-Based Protection of the Hydroxyl Group. chem.harvard.edu
- Kocieński, P.J. (2005) Protecting Groups. 3rd Edition, Thieme, pp. 188–230, ISBN 1588903761.
- Hassner, A.; Reuss, R. H.; Pinnick, H. W. (1975). "Synthetic Methods. VIII. Hydroxylation of Carbonyl Compounds via Silyl Enol Ethers". J. Org. Chem. 40 (23): 3427–3429. doi:10.1021/jo00911a027.
- Rubottom, G. M.; Vazquez, M. A.; Pelegrina, D. R. (1974). "Peracid Oxidation of Trimethylsilyl Enol Ethers: a Facile Α-Hydroxylation Procedure". Tetrahedron Lett. 15 (49–50): 4319–4322. doi:10.1016/S0040-4039(01)92153-7.
- House, H. O.; Gannon, W. F. (1958). "Reaction of β-Diketones with Peracids". J. Org. Chem. 23 (6): 879–884. doi:10.1021/jo01100a030.
- Hubert, A. J.; Starcher, P. S. (1968). "The Baeyer-Villiger Oxidation of Alkyl Oxocyclohexanecarboxylates". J. Chem. Soc. C: 2500. doi:10.1039/j39680002500.
- Kürti, pp. 64–65.
- Li, pp. 68–99.
- Clark, R. D.; Heathcock, C. H. Ozonation of Silylketenes (1974). "Ozonization of silyloxyalkenes". Tetrahedron Lett. 15 (23): 2027–2030. doi:10.1016/S0040-4039(01)82622-8.CS1 maint: multiple names: authors list (link)
- House, H. O.; Czuba, L. J.; Gall, M.; Olmstead, H. D. (1969). "Chemistry of Carbanions. XVIII. Preparation of Trimethylsilyl Enol Ethers". J. Org. Chem. 34 (8): 2324–2336. doi:10.1021/jo01260a018.
- Rubottom, G. M.; Gruber, J. M. (1978). "M-Chloroperbenzoic Acid Oxidation of 2-Trimethylsilyloxy-1, 3-Dienes. Synthesis of. Alpha.-Hydroxy and. Alpha.-Acetoxy Enones". J. Org. Chem. 43 (8): 1599–1602. doi:10.1021/jo00402a030.
- Jauch, J. Stereochemistry of the Rubottom Oxidation with Bicyclic Silyl Enol Ethers; Synthesis and Dimerization Reactions of Bicyclic Α-Hydroxy Ketones (1994). "Stereochemistry of the rubottom oxidation with bicyclic silyl enol ethers; synthesis and dimerization reactions of bicyclic α-hydroxy ketones". Tetrahedron. 50 (45): 12903–12912. doi:10.1016/S0040-4020(01)81209-6.CS1 maint: multiple names: authors list (link)
- Davis, F. A.; Sheppard, A. C. (1987). "Oxidation of Silyl Enol Ethers Using 2-Sulfonyloxaziridines. Synthesis of. Α-Siloxy Epoxides and α-Hydroxy Carbonyl Compounds". J. Org. Chem. 52 (5): 954–955. doi:10.1021/jo00381a051.
- Hashiyama, T.; Morikawa, K.; Sharpless, K. B. (1992). "Alpha.-Hydroxy Ketones in High Enantiomeric Purity From Asymmetric Dihydroxylation of Enol Ethers". J. Org. Chem. 57 (19): 5067–5068. doi:10.1021/jo00045a011.
- Zhu, Y.; Tu, Y.; Yu, H.; Shi, Y. (1998). "Highly Enantioselective Epoxidation of Enol Silyl Ethers and Esters". Tetrahedron Lett. 39 (43): 7819–7822. doi:10.1016/S0040-4039(98)01711-0.
- Adam, W.; Fell, R. T.; Saha-Möller, C. R.; Zhao, C.-G. (1998). "Synthesis of Optically Active Α-Hydroxy Ketones by Enantioselective Oxidation of Silyl Enol Ethers with a Fructose-Derived Dioxirane". Tetrahedron. 9 (3): 397–401. doi:10.1016/S0957-4166(98)00005-6.
- Adam, W.; Fell, R. T.; Stegmann, V. R.; Saha-Möller, C. R. (1998). "Synthesis of Optically Active Α-Hydroxy Carbonyl Compounds by the Catalytic, Enantioselective Oxidation of Silyl Enol Ethers and Ketene Acetals with (Salen) Manganese (III) Complexes". J. Am. Chem. Soc. 120 (4): 708–714. doi:10.1021/ja9726668.
- Stankovic, S.; Espenson, J. H. (1998). "Facile Oxidation of Silyl Enol Ethers with Hydrogen Peroxide Catalyzed by Methyltrioxorhenium". J. Org. Chem. 63 (12): 4129–4130. doi:10.1021/jo972315b.
- Rubottom, G. M.; Gruber, J. M.; Marrero, R.; Juve, H. D., Jr; Kim, C. W. (1983). "Oxidation of Alkyl Trimethylsilyl Ketene Acetals with Lead (IV) Carboxylates". J. Org. Chem. 48 (25): 4940–4944. doi:10.1021/jo00173a031.CS1 maint: multiple names: authors list (link)
- Dayan, S.; Bareket, Y.; Rozen, S. (1999). "An Efficient Α-Hydroxylation of Carbonyls Using the HOF•CH3CN Complex". Tetrahedron. 55 (12): 3657–3664. doi:10.1016/S0040-4020(98)01173-9.
- Still, W. C. (1979). "(±)-Periplanone-B. Total synthesis and structure of the sex excitant pheromone of the American cockroach". J. Am. Chem. Soc. 101 (9): 2493–2495. doi:10.1021/ja00503a048.
- Nicolaou, K.C; Sorensen, E. J (1996) Classics in Total Synthesis: Targets, Strategies, Methods, Wiley, pp. 211–220, ISBN 3527292314.
- Ghosh, A. K.; Li, J. (2009). "An Asymmetric Total Synthesis of Brevisamide". Org. Lett. 11 (18): 4164–4167. doi:10.1021/ol901691d. PMC 2812931. PMID 19694486.
- Dossetter, A. G.; Jamison, T. F.; Jacobsen, E. N. (1999). "Highly Enantio-and Diastereoselective Hetero-Diels-Alder Reactions Catalyzed by New Chiral Tridentate Chromium (III) Catalysts". Angew. Chem. Int. Ed. Engl. 38 (16): 2398–2400. doi:10.1002/(SICI)1521-3773(19990816)38:16<2398::AID-ANIE2398>3.0.CO;2-E. PMID 10458800.
- Wang, H.; Andemichael, Y. W.; Vogt, F. G. (2009). "A Scalable Synthesis of 2 S-Hydroxymutilin via a Modified Rubottom Oxidation". J. Org. Chem. 74 (1): 478–481. doi:10.1021/jo801969e. PMID 19053581.
- Evans, D.A. Chem 206: pKa Table Archived 2013-10-02 at the Wayback Machine. evans.harvard.edu
- Yamaguchi, J.; Hayashi, Y. (2010). "Syntheses of Fumagillin and Ovalicin". Chem. Eur. J. 16 (13): 3884–3901. doi:10.1002/chem.200902433. PMID 20209516.
- Corey, E. J.; Dittami, J. P. (1985). "Total Synthesis of (+/-)-Ovalicin". J. Am. Chem. Soc. 107: 256–257. doi:10.1021/ja00287a049.
- Bath, S.; Billington, D. C.; Gero, S. D.; Quiclet-Sire, B.; Samadi, M. (1994). "Total Synthesis of (−)-Ovalicine From L-Quebrachitol". J. Chem. Soc. Chem. Commun. (12): 1495–1496. doi:10.1039/c39940001495.
- Barton, D. H. R.; Bath, S.; Billington, D. C.; Gero, S. D.; Quiclet-Sire, B. A.; Samadi, M. (1995). "Total Synthesis of (−)-Ovalicin and Analogues From L-Quebrachitol". J. Chem. Soc., Perkin Trans. 1 (12): 1551. doi:10.1039/p19950001551.
- Corey, E. J.; Guzman-Perez, A.; Noe, M. C. (1994). "Short Enantioselective Synthesis of (−)-Ovalicin, a Potent Inhibitor of Angiogenesis, Using Substrate-Enhanced Catalytic Asymmetric Dihydroxylation". J. Am. Chem. Soc. 116 (26): 12109–12110. doi:10.1021/ja00105a084.
- Yadav, J.; Reddy, P.; Reddy, B. (2010). "Stereoselective Total Synthesis of (−)-Ovalicin". Synlett. 2010 (3): 457–461. doi:10.1055/s-0029-1219191.
- Yunes, R. A.; Pizzolatti, M. G.; Sant'Ana, A. E.; Hawkes, G. E.; Calixto, J. B. (1993). "The Structure of Velutinol a, an Anti‐Inflammatory Compound with a Novel Pregnane Skeleton". Phytochemical Analysis. 4 (2): 76–81. doi:10.1002/pca.2800040205.
- Isaka, N.; Tamiya, M.; Hasegawa, A.; Ishiguro, M. (2011). "A Concise Total Synthesis of the Non-Peptide Bradykinin B1 Receptor Antagonist Velutinol A". Eur. J. Org. Chem. 2012: 665–668. doi:10.1002/ejoc.201101728.
- Clive, D. L. J.; Zhang, C. (1995). "Studies on the Degradation of Mevinolin and Compactin: A Formal Route to Semisynthetic Analogues". J. Org. Chem. 60 (5): 1413–1427. doi:10.1021/jo00110a051.
- Reddy, K. K.; Saady, M.; Falck, J. R.; Whited, G. (1995). "Intracellular Mediators: Synthesis of L-α-Phosphatidyl-D-myo-inositol 3,4,5-Trisphosphate and Glyceryl Ether Analogs". J. Org. Chem. 60 (11): 3385–3390. doi:10.1021/jo00116a023.
- Sigma Aldrich mCPBA technical Bulletin
Bibliography
- Kürti, L.; Czakó, B. (2005) Strategic Applications of Named Reactions in Organic Synthesis, Elsevier, ISBN 0124297854.
- Li, J.J. (2009) Name Reactions: A Collection of Detailed Mechanisms and Synthetic Applications, 4th Edition, Springer, ISBN 8132204298