Cascade reaction
A cascade reaction, also known as a domino reaction or tandem reaction, is a chemical process that comprises at least two consecutive reactions such that each subsequent reaction occurs only in virtue of the chemical functionality formed in the previous step.[1] In cascade reactions, isolation of intermediates is not required, as each reaction composing the sequence occurs spontaneously. In the strictest definition of the term, the reaction conditions do not change among the consecutive steps of a cascade and no new reagents are added after the initial step.[1][2] By contrast, one-pot procedures similarly allow at least two reactions to be carried out consecutively without any isolation of intermediates, but do not preclude the addition of new reagents or the change of conditions after the first reaction. Thus, any cascade reaction is also a one-pot procedure, while the reverse does not hold true.[1] Although often composed solely of intramolecular transformations, cascade reactions can also occur intermolecularly, in which case they also fall under the category of multicomponent reactions.[3]

The main benefits of cascade sequences include high atom economy and reduction of waste generated by the several chemical processes, as well as of the time and work required to carry them out.[1][3][4] The efficiency and utility of a cascade reaction can be measured in terms of the number of bonds formed in the overall sequence, the degree of increase in the structural complexity via the process, and its applicability to broader classes of substrates.[2][5]
The earliest example of a cascade reaction is arguably the synthesis of tropinone reported in 1917 by Robinson.[6] Since then, the use of cascade reactions has proliferated in the area of total synthesis. Similarly, the development of cascade-driven organic methodology has also grown tremendously. This increased interest in cascade sequences is reflected by the numerous relevant review articles published in the past couple of decades.[1][2][3][4][5][7][8][9][10] A growing area of focus is the development of asymmetric catalysis of cascade processes by employing chiral organocatalysts or chiral transition-metal complexes.[3][7][10][11]
Classification of cascade reactions is sometimes difficult due to the diverse nature of the many steps in the transformation. K. C. Nicolaou labels the cascades as nucleophilic/electrophilic, radical, pericyclic or transition-metal-catalyzed, based on the mechanism of the steps involved. In the cases in which two or more classes of reaction are included in a cascade, the distinction becomes rather arbitrary and the process is labeled according to what can be arguably considered the “major theme”.[4] In order to highlight the remarkable synthetic utility of cascade reactions, the majority of the examples below come from the total syntheses of complex molecules.
Nucleophilic/electrophilic cascades
Nucleophilic/electrophilic cascades are defined as the cascade sequences in which the key step constitutes a nucleophilic or electrophilic attack.[4]
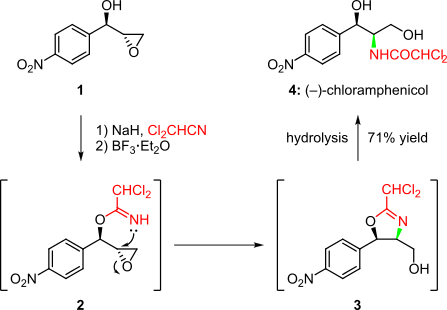
An example of such a cascade is seen in the short enantioselective synthesis of the broad-spectrum antibiotic (–)-chloramphenicol, reported by Rao et al. (Scheme 1).[3][12] Herein, the chiral epoxy-alcohol 1 was first treated with dichloroacetonitrile in the presence of NaH. The resulting intermediate 2 then underwent a BF3·Et2O-mediated cascade reaction. Intramolecular opening of the epoxide ring yielded intermediate 3, which, after an in situ hydrolysis facilitated by excess BF3·Et2O, afforded (–)-chloramphenicol (4) in 71% overall yield.[3][12]
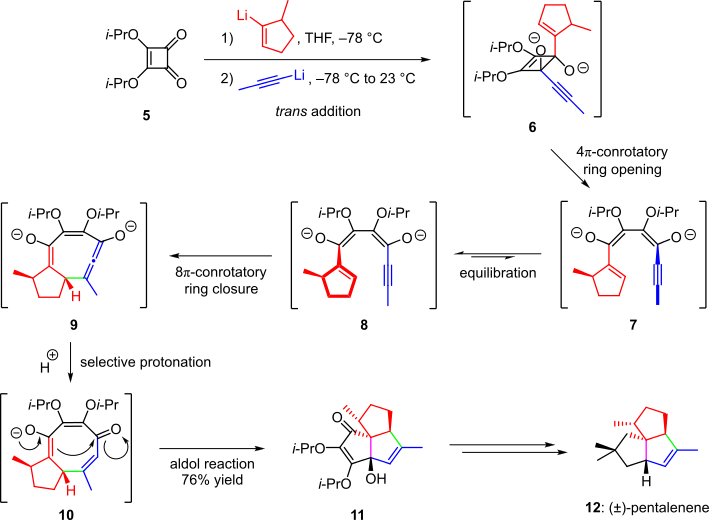
A nucleophilic cascade was also employed in the total synthesis of the natural product pentalenene (Scheme 2).[4][13] In this procedure, squarate ester 5 was treated with (5-methylcyclopent-1-en-1-yl)lithium and propynyllithium. The two nucleophilic attacks occurred predominantly with trans addition to afford intermediate 6, which spontaneously underwent a 4π-conrotatory electrocyclic opening of the cyclobutene ring. The resulting conjugated species 7 equilibrated to conformer 8, which more readily underwent an 8π-conrotatory electrocyclization to the highly strained intermediate 9. The potential to release strain directed protonation of 9 such that species 10 was obtained selectively. The cascade was completed by an intramolecular aldol condensation that afforded product 11 in 76% overall yield. Further elaboration afforded the target (±)-pentalenene (12).[4][13]
Organocatalytic cascades
A subcategory of nucleophilic/electrophilic sequences is constituted by organocatalytic cascades, in which the key nucleophilic attack is driven by organocatalysis.
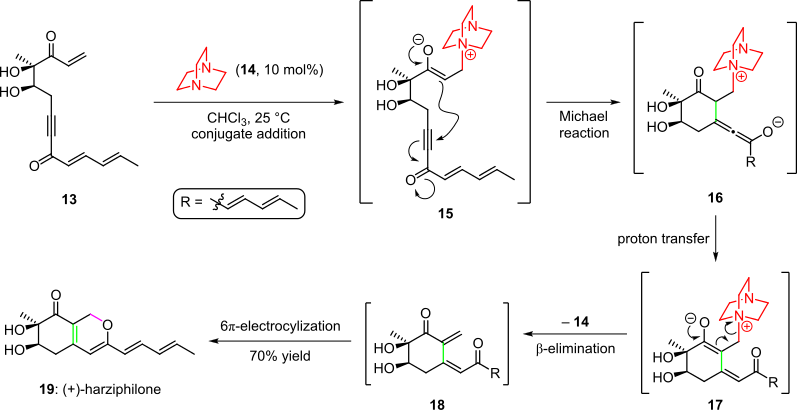
An organocatalytic cascade was employed in the total synthesis of the natural product harziphilone, reported by Sorensen et al. in 2004 (Scheme 3).[4][14] Herein, treatment of the enone starting material 13 with organocatalyst 14 yielded intermediate 15 via conjugate addition. Subsequent cyclization by the intramolecular Michael addition of the enolate into the triple bond of the system gave species 16, which afforded intermediate 17 after proton transfer and tautomerization. The cascade was completed by elimination of the organocatalyst and a spontaneous 6π-electrocyclic ring closure of the resultant cis-dienone 18 to (+)-harziphilone (19) in 70% overall yield.[4][14]
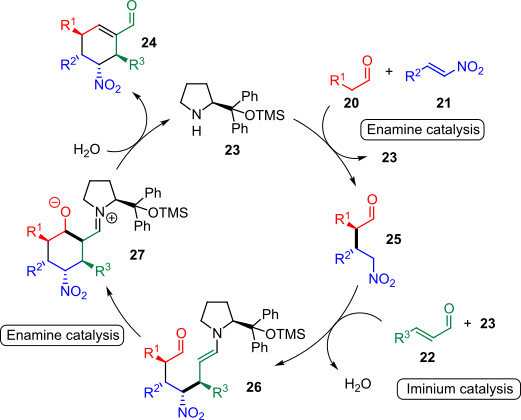
An outstanding triple organocatalytic cascade was reported by Raabe et al. in 2006. Linear aldehydes (20), nitroalkenes (21) and α,β-unsaturated aldehydes (22) could be condensed together organocatalytically to afford tetra-substituted cyclohexane carbaldehydes (24) with moderate to excellent diastereoselectivity and complete enantiocontrol (Scheme 4). The transformation is mediated by the readily available proline-derived organocatalyst 23.[15]

The transformation was proposed to proceed via a Michael addition/Michael addition/aldol condensation sequence (Scheme 5).[15] In the first step, Michael addition of aldehyde 20 to nitroalkene 21 occurs through enamine catalysis, yielding nitroalkane 25. Condensation of α,β-unsaturated aldehyde 22 with the organocatalyst then facilitates the conjugate addition of 25 to give intermediate enamine 26, which is prone to undergo an intramolecular aldol condensation to iminium species 27. Organocatalyst 23 is regenerated by hydrolysis, along with the product 24, thus closing the triple cascade cycle.[15]
Radical cascades
Radical cascades are those in which the key step constitutes a radical reaction. The high reactivity of free radical species renders radical-based synthetic approaches decidedly suitable for cascade reactions.[4]
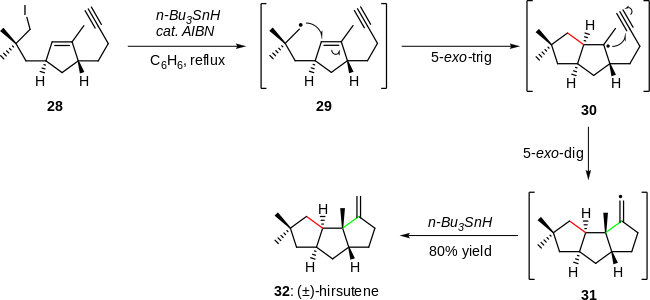
One of the most widely recognized examples of the synthetic utility of radical cascades is the cyclization sequence employed in the total synthesis of (±)-hirsutene, in 1985 (Scheme 6).[4][16] Herein, alkyl iodide 28 was converted to the primary radical intermediate 29, which underwent a 5-exo-trig cyclization to afford reactive species 30. A subsequent 5-exo-dig radical cyclization lead to intermediate 31, which upon quenching gave the target (±)-hirsutene (32) in 80% overall yield.[4][16]
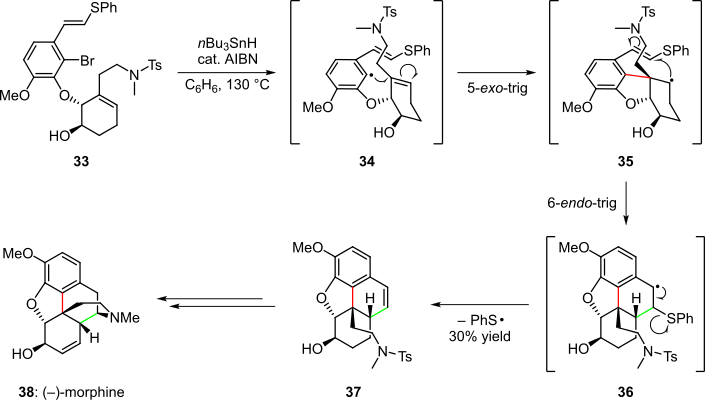
A cascade radical process was also used in one of the total syntheses of (–)-morphine (Scheme 7).[4][17][18] Aryl bromide 33 was converted to the corresponding radical species 34 by treatment with tri-n-butyltin hydride. A 5-exo-trig cyclization then occurred to give intermediate 35 stereoselectively in virtue of the stereochemistry of the ether linkage. In the next step of the cascade, the geometric constraints of 35 forbid the kinetically favored 5-exo-trig cyclization pathway; instead secondary benzylic radical species 36 was obtained via a geometrically-allowed 6-endo-trig cyclization. Subsequent elimination of the phenyl sulfinyl radical afforded product 37 in 30% overall yield, which was further elaborated to (–)-morphine (38).[4][17][18]
Pericyclic cascades
Possibly the most widely encountered kind of process in cascade transformations, pericyclic reactions include cycloadditions, electrocyclic reactions and sigmatropic rearrangements.[4] Although some of the abovementioned instances of nucleophilic/electrophilic and radical cascades involved pericyclic processes, this section contains only cascade sequences that are solely composed of pericyclic reactions or in which such a reaction arguably constitutes the key step.
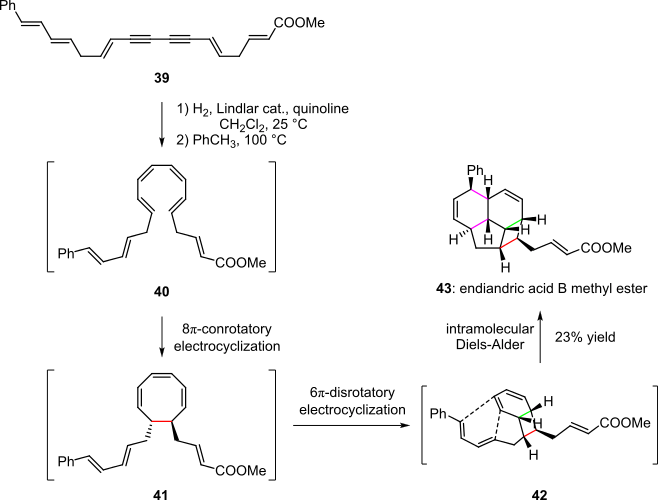
A representative example of a pericyclic cascade is the endiandric acid cascade reported by Nicolaou et al. in 1982 (Scheme 8).[4][19] Herein the highly unsaturated system 39 was first hydrogenated to the conjugated tetraene species 40, which upon heating underwent an 8π-conrotatory electrocyclic ring closure, yielding cyclic intermediate 41. A second spontaneous electrocyclization, this time a 6π-disrotatory ring closure, converted 41 to the bicyclic species 42, the geometry and stereochemistry of which favored a subsequent intramolecular Diels-Alder reaction. The methyl ester of endiandric acid B (43) was thus obtained in 23% overall yield.[4][19]
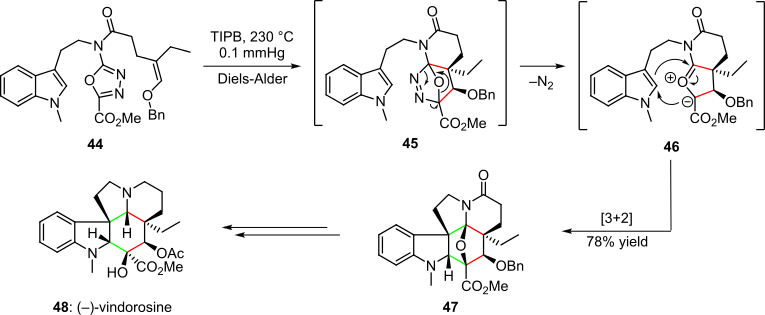
A pericyclic sequence involving intramolecular hetero-cycloaddition reactions was employed in the total synthesis of naturally occurring alkaloid (–)-vindorosine (Scheme 9).[4][20] Rapid access to the target was achieved from a solution of 1,3,4-oxadiazole 44 in triisopropyl benzene subjected to high temperatures and reduced pressure. First an inverse-electron-demand hetero-Diels-Alder reaction occurred to give intermediate 45. Thermodynamically favorable loss of nitrogen generated the 1,3-dipole-containing species 46. A spontaneous intramolecular [3+2] cycloaddition of the 1,3-dipole and the indole system then formed the endo-product 47 in 78% overall yield. Further elaboration yielded the target natural product 48.[4][20]
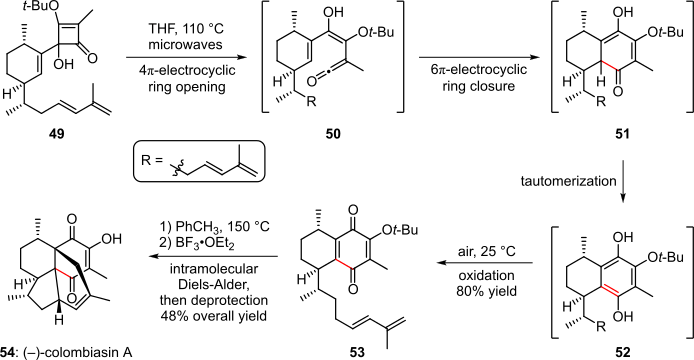
The total synthesis of (–)-colombiasin A reported in 2005 by the Harrowven group included an electrocyclic cascade (Scheme 10).[4][21] When subjected to heat via microwave irradiation, squarate derivative 49 underwent an electrocyclic opening of the cyclobutene ring, followed by a 6π-electrocyclic ring closure that yielded bicyclic intermediate 51. Tautomerization thereof gave the aromatic species 52, which upon exposure to air was oxidized to product 53 in 80% overall yield. The target (–)-colombiasin A (54) was then obtained from 53 via a heat-facilitated Diels-Alder reaction followed by cleavage of the tert-butyl protecting group.[4][21]
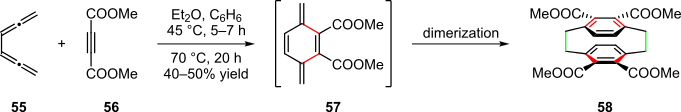
Certain [2,2]paracyclophanes can also be obtained via pericyclic cascades, as reported by the Hopf group in 1981 (Scheme 11).[1][22] In this sequence, a Diels-Alder reaction between 1,2,4,5-hexatetraene 55 and dienophile 56 first formed the highly reactive intermediate 57, which subsequently dimerized to yield [2,2]paracyclophane 58.[1][22]
Transition-metal-catalyzed cascades
Transition-metal-catalyzed cascade sequences combine the novelty and power of organometallic chemistry with the synthetic utility and economy of cascade reactions, providing an even more ecologically and economically desirable approach to organic synthesis.[4]
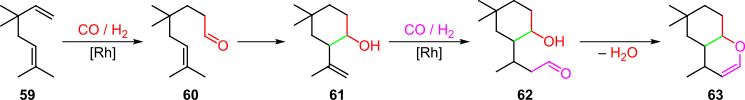
For instance, rhodium catalysis was used to convert acyclic monoterpenes of the type 59 to 4H-chromen products in a hydroformylation cascade (Scheme 12).[8][23] First, selective rhodium-catalyzed hydroformylation of the less sterically hindered olefin bond in 59 yielded unsaturated aldehyde 60, which under the same conditions was then converted to intermediate 61 via a carbonyl-ene reaction. A second rhodium-catalyzed hydroformylation to species 62 was followed by condensation to form 4H-chromen products of the type 63 in 40% overall yield.[8][23]
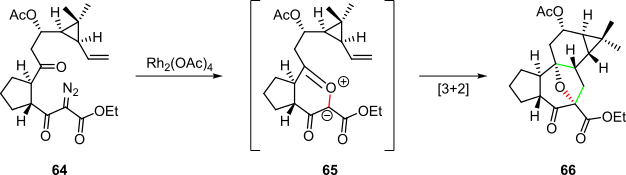
Rhodium catalysis was also employed to initiate a cyclization/cycloaddition cascade in the synthesis of a tigliane reported by the Dauben group (Scheme 13).[2][24] Treatment of diazoimide 64 with rhodium(II) acetate dimer generated a carbenoid that yielded reactive ylide 65 after an intramolecular cyclization with the neighboring carbonyl group. An intramolecular [3+2] cycloaddition then spontaneously occurred to afford the target tigliane 66.[2][24]
The formal intramolecular [4+2] cycloaddition of 1,6-enynes of the type 67 mediated by gold catalysis is another example of a transition-metal-catalyzed cascade (Scheme 14).[25][26] A variety of 1,6-enynes reacted under mild conditions in the presence of Au(I) complexes 68a–b to yield the tricyclic products 69 in moderate to excellent yields.[25][26]
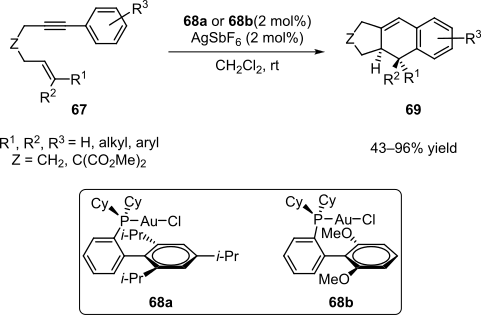
This formal cycloaddition was proposed to proceed via the cascade process shown in Scheme 15.[25][26] Complexation of the 1,6-enyne 67 with the cationic form of the catalyst yields intermediate 70, in which the activated triple bond is attacked by the olefin functionality to yield substituted cyclopropane 71. Electrophilic opening of the three-membered ring forms cationic species 72, which undergoes a Friedel-Crafts-type reaction and then rearomatizes to give tricyclic product 69.[25][26] Due to the nature of the interaction of gold complexes with unsaturated systems, this process could also be considered an electrophilic cascade.
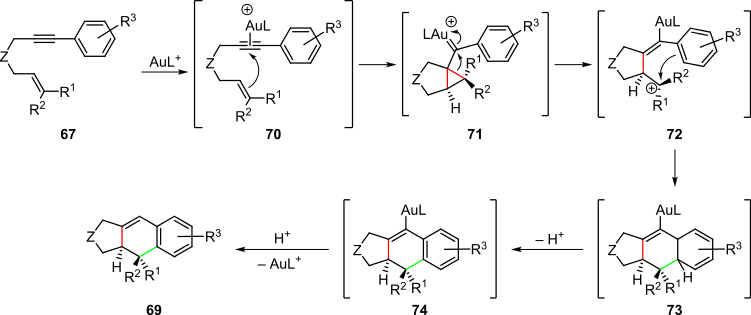
An example of palladium-catalyzed cascades is represented by the asymmetric polyene Heck cyclization used in the preparation of (+)-xestoquinone from triflate substrate 75 (Scheme 16).[4][27] Oxidative addition of the aryl–triflate bond into the palladium(0) complex in the presence of chiral diphosphine ligand (S)-binap yields chiral palladium(II) complex 77. This step is followed by the dissociation of the triflate anion, association of the neighboring olefin and 1,2-insertion of the naphthyl group into the olefin to yield intermediate 79. A second migratory insertion into the remaining olefin group followed by a β-elimination then occurs to afford product 81 in 82% overall yield and with moderate enantioselectivity. The palladium(0) catalyst is also regenerated in this step, thus allowing the cascade to be reinitiated.[4][27]
Multistep tandem reactions
Multistep tandem reactions (or cascade reactions) are a sequence of chemical transformations (usually more than two steps) that happens consecutively to convert a starting material to a complex product.[28] This kind of organic reactions are designed to construct difficult structures encountered in natural product total synthesis.
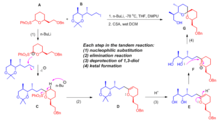
In the total synthesis of spiroketal ionophore antibiotic routiennocin 1 (Fig. 1), the central spiroketal skeleton was constructed by a multistep tandem reaction (Fig. 2).[29] Fragment A and fragment B were coupled in a single step to form the key intermediate G that could be further elaborated to afford the final product routiennocin.
Four chemical transformations happened in this tandem reaction. First, treating fragment A with n-butyllithium formed carbon anion that attacked the alkyliodide part of fragment B to generate intermediate C (step 1). Then a 3, 4-dihydropyran derivative D was formed through base-mediated elimination reaction on intermediate C (step 2). The protecting group on 1, 3-diol moiety in intermediate D was removed by acid treatment to give the diol product E (step 3). The spiroketal product G was generated via intramolecular ketal formation reaction. This multistep tandem reaction greatly simplified the construction of this complex spiroketal structure and eased the path towards the total synthesis of routiennocin.
References
- Tietze, L. F.; Beifuss, U. Angew. Chem. Int. Ed. 1993, 32, 131–163.
- Padwa, A.; Bur, S. K. Tetrahedron 2007, 63, 5341–5378.
- Pellissier, H. Tetrahedron 2006, 62, 1619–1665.
- Nicolaou, K. C.; Edmonds, D. J.; Bulger, P. G. Angew. Chem. Int. Ed. 2006, 45, 7134–7186.
- Tietze, L. F. Chem. Rev. 1996, 96, 115–136.
- Robinson, R. J. Chem. Soc. Trans. 1917, 111, 762.
- Pellissier, H. Tetrahedron 2006, 62, 2143–2173.
- Wasilke, J. C.; Obrey, S. J.; Baker, R. T.; Bazan, G. C. Chem. Rev. 2005, 105, 1001–1020.
- Chapman, C.; Frost, C. Synthesis (Stuttg). 2007, 2007, 1–21.
- Enders, D.; Grondal, C.; Hüttl, M. R. M. Angew. Chem. Int. Ed. 2007, 46, 1570–1581.
- Grondal, C.; Jeanty, M.; Enders, D. Nat. Chem. 2010, 2, 167–178.
- Bhaskar, G.; Satish Kumar, V.; Venkateswara Rao, B. Tetrahedron: Asymmetry 2004, 15, 1279–1283.
- Paquette, L. A.; Geng, F. Org. Lett. 2002, 4, 4547–4549.
- Stark, L. M.; Pekari, K.; Sorensen, E. J. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 12064–12066.
- Enders, D.; Hüttl, M. R. M.; Grondal, C.; Raabe, G. Nature 2006, 441, 861–863.
- Curran, D. P.; Chen, M.-H. Tetrahedron Lett. 1985, 26, 4991–4994.
- Parker, K. A.; Fokas, D. J. Am. Chem. Soc. 1992, 114, 9688–9689.
- Parker, K. A.; Fokas, D. J. Org. Chem. 2006, 71, 449–455.
- Nicolaou, K. C.; Petasis, N. A.; Zipkin, R. E.; Uenishi, J. J. Am. Chem. Soc. 1982, 104, 5555–5557.
- Elliott, G. I.; Velcicky, J.; Ishikawa, H.; Li, Y.; Boger, D. L. Angew. Chem. Int. Ed. 2006, 45, 620–622.
- Harrowven, D. C.; Pascoe, D. D.; Demurtas, D.; Bourne, H. O. Angew. Chem. Int. Ed. 2005, 44, 1221–1222.
- Hopf, H.; Bohm, I.; Kleinschroth, J. Org. Synth. 1981, 60, 41.
- Roggenbuck, R.; Eilbracht, P. Tetrahedron Lett. 1999, 40, 7455–7456.
- Dauben, W. G.; Dinges, J.; Smith, T. C. J. Org. Chem. 1993, 58, 7635–7637.
- Jiménez-Núñez, E.; Echavarren, A. M. Chem. Rev. 2008, 108, 3326.
- Nieto-Oberhuber, C.; López, S.; Echavarren, A. M. J. Am. Chem. Soc. 2005, 127, 6178–6179.
- Maddaford, S. P.; Andersen, N. G.; Cristofoli, W. A.; Keay, B. A. J. Am. Chem. Soc. 1996, 118, 10766–10773.
- Nicolaou, K. C.; Edmonds, David J.; Bulger, Paul G. Angew. Chem. Int. Ed. 2006, 45, 7134-7186.
- Diez-Martin, D. Kotecha, N. R.; Ley, S. V.; Mantegani, S.; Menendez, J. C.; Organ, H. M.; White, A. D., Tetrahedron, 1992, 48, 1899-7938.
External links
| Wikiquote has quotations related to: Cascade reaction |
- Chemical Knots at The Periodic Table of Videos (University of Nottingham)