Lisdexamfetamine
Lisdexamfetamine, sold under the brand name Vyvanse among others, is a medication that is a derivative of amphetamine. It is mainly used to treat attention deficit hyperactivity disorder (ADHD) in people over the age of five as well as moderate-to-severe binge eating disorder in adults.[1] Lisdexamfetamine is taken by mouth.[1][6] In the United Kingdom it is usually less preferred than methylphenidate.[7] Its effects generally begin within 2 hours and last for up to 12 hours.[1]
 | |
 | |
| Clinical data | |
|---|---|
| Trade names | Tyvense, Elvanse, Vyvanse, others |
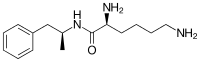
| Other names | (2S)-2,6-Diamino-N-[(2S)-1-phenylpropan-2-yl]hexanamide N-[(2S)-1-Phenyl-2-propanyl]-L-lysinamide |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a607047 |
| License data |
|
| Pregnancy category | |
| Dependence liability | High[1][2] |
| Addiction liability | Moderate |
| Routes of administration | By mouth (capsules) |
| ATC code | |
| Legal status | |
| Legal status |
|
| Pharmacokinetic data | |
| Bioavailability | 96.4%[3] |
| Metabolism | Hydrolysis by enzymes in red blood cells initially. Subsequent metabolism follows Amphetamine#Pharmacokinetics. |
| Onset of action | 2 h[4][5] |
| Elimination half-life | ≤1 h (prodrug molecule) 9–11 h (dextroamphetamine) |
| Duration of action | 10–12 h[2][4][5] |
| Excretion | Renal: ~2% |
| Identifiers | |
| |
| CAS Number |
|
| PubChem CID | |
| IUPHAR/BPS | |
| DrugBank | |
| ChemSpider | |
| UNII | |
| ChEMBL | |
| CompTox Dashboard (EPA) | |
| Chemical and physical data | |
| Formula | C15H25N3O |
| Molar mass | 263.385 g·mol−1 |
| 3D model (JSmol) | |
| |
| |
| | |
Common side effects of lisdexamfetamine include loss of appetite, anxiety, diarrhea, trouble sleeping, irritability, and nausea.[1] Rare but serious side effects include mania, sudden cardiac death in those with underlying heart problems, and psychosis.[1] It has a high potential for abuse per the DEA.[1][6] Serotonin syndrome may occur if used with certain other medications.[1] Its use during pregnancy may result in harm to the baby and use during breastfeeding is not recommended by the manufacturer.[7][1][6] Lisdexamfetamine is a central nervous system (CNS) stimulant that works after being converted by the body into dextroamphetamine.[1][8] Chemically, lisdexamfetamine is composed of the amino acid L-lysine, attached to dextroamphetamine.[9]
Lisdexamfetamine was approved for medical use in the United States in 2007.[1] A month's supply in the United Kingdom costs the British National Health Service about £58 as of 2019.[7] In the United States, the wholesale cost of this amount is about US$264.[10] In 2017, it was the 91st most commonly prescribed medication in the United States, with more than eight million prescriptions.[11][12] It is a Schedule II controlled substance in the United Kingdom and a Schedule II controlled substance in the United States.[7][13]
Uses
Medical
Lisdexamfetamine is used primarily as a treatment for attention deficit hyperactivity disorder (ADHD) and binge eating disorder;[14] it has similar off-label uses as those of other pharmaceutical amphetamines.[2] Individuals over the age of 65 were not commonly tested in clinical trials of lisdexamfetamine for ADHD.[14] Long-term amphetamine exposure at sufficiently high doses in some animal species is known to produce abnormal dopamine system development or nerve damage,[15][16] but, in humans with ADHD, pharmaceutical amphetamines, at therapeutic dosages, appear to improve brain development and nerve growth.[17][18][19] Reviews of magnetic resonance imaging (MRI) studies suggest that long-term treatment with amphetamine decreases abnormalities in brain structure and function found in subjects with ADHD, and improves function in several parts of the brain, such as the right caudate nucleus of the basal ganglia.[17][18][19]
Reviews of clinical stimulant research have established the safety and effectiveness of long-term continuous amphetamine use for the treatment of ADHD.[20][21][22] Randomized controlled trials of continuous stimulant therapy for the treatment of ADHD spanning 2 years have demonstrated treatment effectiveness and safety.[20][21] Two reviews have indicated that long-term continuous stimulant therapy for ADHD is effective for reducing the core symptoms of ADHD (i.e., hyperactivity, inattention, and impulsivity), enhancing quality of life and academic achievement, and producing improvements in a large number of functional outcomes[note 1] across 9 categories of outcomes related to academics, antisocial behavior, driving, non-medicinal drug use, obesity, occupation, self-esteem, service use (i.e., academic, occupational, health, financial, and legal services), and social function.[20][22] One review highlighted a nine-month randomized controlled trial of amphetamine treatment for ADHD in children that found an average increase of 4.5 IQ points, continued increases in attention, and continued decreases in disruptive behaviors and hyperactivity.[21] Another review indicated that, based upon the longest follow-up studies conducted to date, lifetime stimulant therapy that begins during childhood is continuously effective for controlling ADHD symptoms and reduces the risk of developing a substance use disorder as an adult.[20]
Current models of ADHD suggest that it is associated with functional impairments in some of the brain's neurotransmitter systems;[23] these functional impairments involve impaired dopamine neurotransmission in the mesocorticolimbic projection and norepinephrine neurotransmission in the noradrenergic projections from the locus coeruleus to the prefrontal cortex.[23] Psychostimulants like methylphenidate and amphetamine are effective in treating ADHD because they increase neurotransmitter activity in these systems.[24][23][25] Approximately 80% of those who use these stimulants see improvements in ADHD symptoms.[26] Children with ADHD who use stimulant medications generally have better relationships with peers and family members, perform better in school, are less distractible and impulsive, and have longer attention spans.[27][28] The Cochrane reviews[note 2] on the treatment of ADHD in children, adolescents, and adults with pharmaceutical amphetamines stated that short-term studies have demonstrated that these drugs decrease the severity of symptoms, but they have higher discontinuation rates than non-stimulant medications due to their adverse side effects.[30][31] A Cochrane review on the treatment of ADHD in children with tic disorders such as Tourette syndrome indicated that stimulants in general do not make tics worse, but high doses of dextroamphetamine could exacerbate tics in some individuals.[32]
Enhancing performance
Cognitive performance
In 2015, a systematic review and a meta-analysis of high quality clinical trials found that, when used at low (therapeutic) doses, amphetamine produces modest yet unambiguous improvements in cognition, including working memory, long-term episodic memory, inhibitory control, and some aspects of attention, in normal healthy adults;[33][34] these cognition-enhancing effects of amphetamine are known to be partially mediated through the indirect activation of both dopamine receptor D1 and adrenoceptor α2 in the prefrontal cortex.[24][33] A systematic review from 2014 found that low doses of amphetamine also improve memory consolidation, in turn leading to improved recall of information.[35] Therapeutic doses of amphetamine also enhance cortical network efficiency, an effect which mediates improvements in working memory in all individuals.[24][36] Amphetamine and other ADHD stimulants also improve task saliency (motivation to perform a task) and increase arousal (wakefulness), in turn promoting goal-directed behavior.[24][37][38] Stimulants such as amphetamine can improve performance on difficult and boring tasks and are used by some students as a study and test-taking aid.[24][38][39] Based upon studies of self-reported illicit stimulant use, 5–35% of college students use diverted ADHD stimulants, which are primarily used for enhancement of academic performance rather than as recreational drugs.[40][41][42] However, high amphetamine doses that are above the therapeutic range can interfere with working memory and other aspects of cognitive control.[24][38]
Physical performance
Amphetamine is used by some athletes for its psychological and athletic performance-enhancing effects, such as increased endurance and alertness;[43][44] however, non-medical amphetamine use is prohibited at sporting events that are regulated by collegiate, national, and international anti-doping agencies.[45][46] In healthy people at oral therapeutic doses, amphetamine has been shown to increase muscle strength, acceleration, athletic performance in anaerobic conditions, and endurance (i.e., it delays the onset of fatigue), while improving reaction time.[43][47][48] Amphetamine improves endurance and reaction time primarily through reuptake inhibition and release of dopamine in the central nervous system.[47][48][49] Amphetamine and other dopaminergic drugs also increase power output at fixed levels of perceived exertion by overriding a "safety switch", allowing the core temperature limit to increase in order to access a reserve capacity that is normally off-limits.[48][50][51] At therapeutic doses, the adverse effects of amphetamine do not impede athletic performance;[43][47] however, at much higher doses, amphetamine can induce effects that severely impair performance, such as rapid muscle breakdown and elevated body temperature.[52][47]
Contraindications
Pharmaceutical lisdexamfetamine dimesylate is contraindicated in patients with hypersensitivity to amphetamine products or any of the formulation's inactive ingredients.[14] It is also contraindicated in patients who have used a monoamine oxidase inhibitor (MAOI) within the last 14 days.[14][53] Amphetamine products are contraindicated by the United States Food and Drug Administration (USFDA) in people with a history of drug abuse, heart disease, or severe agitation or anxiety, or in those currently experiencing arteriosclerosis, glaucoma, hyperthyroidism, or severe hypertension.[54] The USFDA advises anyone with bipolar disorder, depression, elevated blood pressure, liver or kidney problems, mania, psychosis, Raynaud's phenomenon, seizures, thyroid problems, tics, or Tourette syndrome to monitor their symptoms while taking amphetamine.[54] Amphetamine is classified in US pregnancy category C.[54] This means that detriments to the fetus have been observed in animal studies and adequate human studies have not been conducted; amphetamine may still be prescribed to pregnant women if the potential benefits outweigh the risks.[55] Amphetamine has also been shown to pass into breast milk, so the USFDA advises mothers to avoid breastfeeding when using it.[54] Due to the potential for stunted growth, the USFDA advises monitoring the height and weight of children and adolescents prescribed amphetamines.[54] Prescribing information approved by the Australian Therapeutic Goods Administration further contraindicates anorexia.[56]
Adverse effects
Products containing lisdexamfetamine have a comparable drug safety profile to those containing amphetamine.[9]
Physical
Cardiovascular side effects can include hypertension or hypotension from a vasovagal response, Raynaud's phenomenon (reduced blood flow to the hands and feet), and tachycardia (increased heart rate).[52][44][57] Sexual side effects in males may include erectile dysfunction, frequent erections, or prolonged erections.[52] Gastrointestinal side effects may include abdominal pain, constipation, diarrhea, and nausea.[58][52] Other potential physical side effects include appetite loss, blurred vision, dry mouth, excessive grinding of the teeth, nosebleed, profuse sweating, rhinitis medicamentosa (drug-induced nasal congestion), reduced seizure threshold, tics (a type of movement disorder), and weight loss.[sources 1] Dangerous physical side effects are rare at typical pharmaceutical doses.[44]
Amphetamine stimulates the medullary respiratory centers, producing faster and deeper breaths.[44] In a normal person at therapeutic doses, this effect is usually not noticeable, but when respiration is already compromised, it may be evident.[44] Amphetamine also induces contraction in the urinary bladder sphincter, the muscle which controls urination, which can result in difficulty urinating.[44] This effect can be useful in treating bed wetting and loss of bladder control.[44] The effects of amphetamine on the gastrointestinal tract are unpredictable.[44] If intestinal activity is high, amphetamine may reduce gastrointestinal motility (the rate at which content moves through the digestive system);[44] however, amphetamine may increase motility when the smooth muscle of the tract is relaxed.[44] Amphetamine also has a slight analgesic effect and can enhance the pain relieving effects of opioids.[58][44]
USFDA-commissioned studies from 2011 indicate that in children, young adults, and adults there is no association between serious adverse cardiovascular events (sudden death, heart attack, and stroke) and the medical use of amphetamine or other ADHD stimulants.[sources 2] However, amphetamine pharmaceuticals are contraindicated in individuals with cardiovascular disease.[sources 3]
Psychological
At normal therapeutic doses, the most common psychological side effects of amphetamine include increased alertness, apprehension, concentration, initiative, self-confidence and sociability, mood swings (elated mood followed by mildly depressed mood), insomnia or wakefulness, and decreased sense of fatigue.[52][44] Less common side effects include anxiety, change in libido, grandiosity, irritability, repetitive or obsessive behaviors, and restlessness;[sources 4] these effects depend on the user's personality and current mental state.[44] Amphetamine psychosis (e.g., delusions and paranoia) can occur in heavy users.[52][67][68] Although very rare, this psychosis can also occur at therapeutic doses during long-term therapy.[52][68][69] According to the USFDA, "there is no systematic evidence" that stimulants produce aggressive behavior or hostility.[52]
Amphetamine has also been shown to produce a conditioned place preference in humans taking therapeutic doses,[30][70] meaning that individuals acquire a preference for spending time in places where they have previously used amphetamine.[70][71]
Reinforcement disorders
Addiction
| Addiction and dependence glossary[71][72][73][74] | |
|---|---|
| |
| Transcription factor glossary | |
|---|---|
| |
Signaling cascade in the nucleus accumbens that results in amphetamine addiction
|
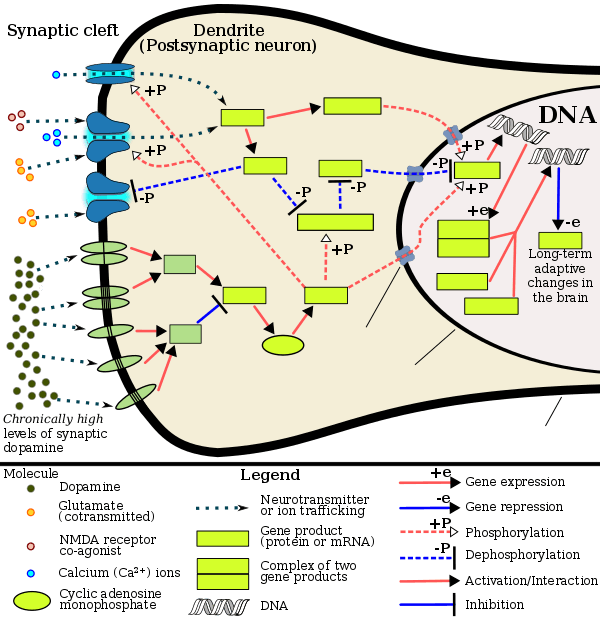
Addiction is a serious risk with heavy recreational amphetamine use, but is unlikely to occur from long-term medical use at therapeutic doses;[20][82][83] in fact, lifetime stimulant therapy for ADHD that begins during childhood reduces the risk of developing substance use disorders as an adult.[20] Compared to other amphetamine pharmaceuticals, lisdexamfetamine may have a lower liability for abuse as a recreational drug.[84] Pathological overactivation of the mesolimbic pathway, a dopamine pathway that connects the ventral tegmental area to the nucleus accumbens, plays a central role in amphetamine addiction.[85][86] Individuals who frequently self-administer high doses of amphetamine have a high risk of developing an amphetamine addiction, since chronic use at high doses gradually increase the level of accumbal ΔFosB, a "molecular switch" and "master control protein" for addiction.[72][87][88] Once nucleus accumbens ΔFosB is sufficiently overexpressed, it begins to increase the severity of addictive behavior (i.e., compulsive drug-seeking) with further increases in its expression.[87][89] While there are currently no effective drugs for treating amphetamine addiction, regularly engaging in sustained aerobic exercise appears to reduce the risk of developing such an addiction.[90][91] Sustained aerobic exercise on a regular basis also appears to be an effective treatment for amphetamine addiction;[sources 5] exercise therapy improves clinical treatment outcomes and may be used as an adjunct therapy with behavioral therapies for addiction.[90][92]
Biomolecular mechanisms
Chronic use of amphetamine at excessive doses causes alterations in gene expression in the mesocorticolimbic projection, which arise through transcriptional and epigenetic mechanisms.[88][93][94] The most important transcription factors[note 3] that produce these alterations are Delta FBJ murine osteosarcoma viral oncogene homolog B (ΔFosB), cAMP response element binding protein (CREB), and nuclear factor-kappa B (NF-κB).[88] ΔFosB is the most significant biomolecular mechanism in addiction because ΔFosB overexpression (i.e., an abnormally high level of gene expression which produces a pronounced gene-related phenotype) in the D1-type medium spiny neurons in the nucleus accumbens is necessary and sufficient[note 4] for many of the neural adaptations and regulates multiple behavioral effects (e.g., reward sensitization and escalating drug self-administration) involved in addiction.[72][87][88] Once ΔFosB is sufficiently overexpressed, it induces an addictive state that becomes increasingly more severe with further increases in ΔFosB expression.[72][87] It has been implicated in addictions to alcohol, cannabinoids, cocaine, methylphenidate, nicotine, opioids, phencyclidine, propofol, and substituted amphetamines, among others.[sources 6]
ΔJunD, a transcription factor, and G9a, a histone methyltransferase enzyme, both oppose the function of ΔFosB and inhibit increases in its expression.[72][88][98] Sufficiently overexpressing ΔJunD in the nucleus accumbens with viral vectors can completely block many of the neural and behavioral alterations seen in chronic drug abuse (i.e., the alterations mediated by ΔFosB).[88] Similarly, accumbal G9a hyperexpression results in markedly increased histone 3 lysine residue 9 dimethylation (H3K9me2) and blocks the induction of ΔFosB-mediated neural and behavioral plasticity by chronic drug use,[sources 7] which occurs via H3K9me2-mediated repression of transcription factors for ΔFosB and H3K9me2-mediated repression of various ΔFosB transcriptional targets (e.g., CDK5).[88][98][99] ΔFosB also plays an important role in regulating behavioral responses to natural rewards, such as palatable food, sex, and exercise.[89][88][102] Since both natural rewards and addictive drugs induce the expression of ΔFosB (i.e., they cause the brain to produce more of it), chronic acquisition of these rewards can result in a similar pathological state of addiction.[89][88] Consequently, ΔFosB is the most significant factor involved in both amphetamine addiction and amphetamine-induced sexual addictions, which are compulsive sexual behaviors that result from excessive sexual activity and amphetamine use.[89][103][104] These sexual addictions are associated with a dopamine dysregulation syndrome which occurs in some patients taking dopaminergic drugs.[89][102]
The effects of amphetamine on gene regulation are both dose- and route-dependent.[94] Most of the research on gene regulation and addiction is based upon animal studies with intravenous amphetamine administration at very high doses.[94] The few studies that have used equivalent (weight-adjusted) human therapeutic doses and oral administration show that these changes, if they occur, are relatively minor.[94] This suggests that medical use of amphetamine does not significantly affect gene regulation.[94]
Pharmacological treatments
As of December 2019, there is no effective pharmacotherapy for amphetamine addiction.[105][106][107] Reviews from 2015 and 2016 indicated that TAAR1-selective agonists have significant therapeutic potential as a treatment for psychostimulant addictions;[108][109] however, as of February 2016, the only compounds which are known to function as TAAR1-selective agonists are experimental drugs.[108][109] Amphetamine addiction is largely mediated through increased activation of dopamine receptors and co-localized NMDA receptors[note 5] in the nucleus accumbens;[86] magnesium ions inhibit NMDA receptors by blocking the receptor calcium channel.[86][110] One review suggested that, based upon animal testing, pathological (addiction-inducing) psychostimulant use significantly reduces the level of intracellular magnesium throughout the brain.[86] Supplemental magnesium[note 6] treatment has been shown to reduce amphetamine self-administration (i.e., doses given to oneself) in humans, but it is not an effective monotherapy for amphetamine addiction.[86]
A systematic review and meta-analysis from 2019 assessed the efficacy of 17 different pharmacotherapies used in RCTs for amphetamine and methamphetamine addiction;[106] it found only low-strength evidence that methylphenidate might reduce amphetamine or methamphetamine self-administration.[106] There was low- to moderate-strength evidence of no benefit for most of the other medications used in RCTs, which included antidepressants (bupropion, mirtazapine, sertraline), antipsychotics (aripiprazole), anticonvulsants (topiramate, baclofen, gabapentin), naltrexone, varenicline, citicoline, ondansetron, prometa, riluzole, atomoxetine, dextroamphetamine, and modafinil.[106]
Behavioral treatments
A 2018 systematic review and network meta-analysis of 50 trials involving 12 different psychosocial interventions for amphetamine, methamphetamine, or cocaine addiction found that combination therapy with both contingency management and community reinforcement approach had the highest efficacy (i.e., abstinence rate) and acceptability (i.e., lowest dropout rate).[111] Other treatment modalities examined in the analysis included monotherapy with contingency management or community reinforcement approach, cognitive behavioral therapy, 12-step programs, non-contingent reward-based therapies, psychodynamic therapy, and other combination therapies involving these.[111]
Additionally, research on the neurobiological effects of physical exercise suggests that daily aerobic exercise, especially endurance exercise (e.g., marathon running), prevents the development of drug addiction and is an effective adjunct therapy (i.e., a supplemental treatment) for amphetamine addiction.[sources 5] Exercise leads to better treatment outcomes when used as an adjunct treatment, particularly for psychostimulant addictions.[90][92][112] In particular, aerobic exercise decreases psychostimulant self-administration, reduces the reinstatement (i.e., relapse) of drug-seeking, and induces increased dopamine receptor D2 (DRD2) density in the striatum.[89][112] This is the opposite of pathological stimulant use, which induces decreased striatal DRD2 density.[89] One review noted that exercise may also prevent the development of a drug addiction by altering ΔFosB or c-Fos immunoreactivity in the striatum or other parts of the reward system.[91]
| Form of neuroplasticity or behavioral plasticity |
Type of reinforcer | Sources | |||||
|---|---|---|---|---|---|---|---|
| Opiates | Psychostimulants | High fat or sugar food | Sexual intercourse | Physical exercise (aerobic) |
Environmental enrichment | ||
| ΔFosB expression in nucleus accumbens D1-type MSNs |
↑ | ↑ | ↑ | ↑ | ↑ | ↑ | [89] |
| Behavioral plasticity | |||||||
| Escalation of intake | Yes | Yes | Yes | [89] | |||
| Psychostimulant cross-sensitization |
Yes | Not applicable | Yes | Yes | Attenuated | Attenuated | [89] |
| Psychostimulant self-administration |
↑ | ↑ | ↓ | ↓ | ↓ | [89] | |
| Psychostimulant conditioned place preference |
↑ | ↑ | ↓ | ↑ | ↓ | ↑ | [89] |
| Reinstatement of drug-seeking behavior | ↑ | ↑ | ↓ | ↓ | [89] | ||
| Neurochemical plasticity | |||||||
| CREB phosphorylation in the nucleus accumbens |
↓ | ↓ | ↓ | ↓ | ↓ | [89] | |
| Sensitized dopamine response in the nucleus accumbens |
No | Yes | No | Yes | [89] | ||
| Altered striatal dopamine signaling | ↓DRD2, ↑DRD3 | ↑DRD1, ↓DRD2, ↑DRD3 | ↑DRD1, ↓DRD2, ↑DRD3 | ↑DRD2 | ↑DRD2 | [89] | |
| Altered striatal opioid signaling | No change or ↑μ-opioid receptors | ↑μ-opioid receptors ↑κ-opioid receptors | ↑μ-opioid receptors | ↑μ-opioid receptors | No change | No change | [89] |
| Changes in striatal opioid peptides | ↑dynorphin No change: enkephalin | ↑dynorphin | ↓enkephalin | ↑dynorphin | ↑dynorphin | [89] | |
| Mesocorticolimbic synaptic plasticity | |||||||
| Number of dendrites in the nucleus accumbens | ↓ | ↑ | ↑ | [89] | |||
| Dendritic spine density in the nucleus accumbens |
↓ | ↑ | ↑ | [89] | |||
Dependence and withdrawal
Drug tolerance develops rapidly in amphetamine abuse (i.e., recreational amphetamine use), so periods of extended abuse require increasingly larger doses of the drug in order to achieve the same effect.[113][114] According to a Cochrane review on withdrawal in individuals who compulsively use amphetamine and methamphetamine, "when chronic heavy users abruptly discontinue amphetamine use, many report a time-limited withdrawal syndrome that occurs within 24 hours of their last dose."[115] This review noted that withdrawal symptoms in chronic, high-dose users are frequent, occurring in roughly 88% of cases, and persist for 3–4 weeks with a marked "crash" phase occurring during the first week.[115] Amphetamine withdrawal symptoms can include anxiety, drug craving, depressed mood, fatigue, increased appetite, increased movement or decreased movement, lack of motivation, sleeplessness or sleepiness, and lucid dreams.[115] The review indicated that the severity of withdrawal symptoms is positively correlated with the age of the individual and the extent of their dependence.[115] Mild withdrawal symptoms from the discontinuation of amphetamine treatment at therapeutic doses can be avoided by tapering the dose.[58]
Overdose
An amphetamine overdose can lead to many different symptoms, but is rarely fatal with appropriate care.[58][53][116] The severity of overdose symptoms increases with dosage and decreases with drug tolerance to amphetamine.[44][53] Tolerant individuals have been known to take as much as 5 grams of amphetamine in a day, which is roughly 100 times the maximum daily therapeutic dose.[53] Symptoms of a moderate and extremely large overdose are listed below; fatal amphetamine poisoning usually also involves convulsions and coma.[52][44] In 2013, overdose on amphetamine, methamphetamine, and other compounds implicated in an "amphetamine use disorder" resulted in an estimated 3,788 deaths worldwide (3,425–4,145 deaths, 95% confidence).[note 7][117]
| System | Minor or moderate overdose[52][44][53] | Severe overdose[sources 8] |
|---|---|---|
| Cardiovascular |
|
|
| Central nervous system |
|
|
| Musculoskeletal |
| |
| Respiratory |
|
|
| Urinary |
|
|
| Other |
|
|
Toxicity
In rodents and primates, sufficiently high doses of amphetamine cause dopaminergic neurotoxicity, or damage to dopamine neurons, which is characterized by dopamine terminal degeneration and reduced transporter and receptor function.[120][121] There is no evidence that amphetamine is directly neurotoxic in humans.[122][123] However, large doses of amphetamine may indirectly cause dopaminergic neurotoxicity as a result of hyperpyrexia, the excessive formation of reactive oxygen species, and increased autoxidation of dopamine.[sources 9] Animal models of neurotoxicity from high-dose amphetamine exposure indicate that the occurrence of hyperpyrexia (i.e., core body temperature ≥ 40 °C) is necessary for the development of amphetamine-induced neurotoxicity.[121] Prolonged elevations of brain temperature above 40 °C likely promote the development of amphetamine-induced neurotoxicity in laboratory animals by facilitating the production of reactive oxygen species, disrupting cellular protein function, and transiently increasing blood–brain barrier permeability.[121]
Psychosis
An amphetamine overdose can result in a stimulant psychosis that may involve a variety of symptoms, such as delusions and paranoia.[67][68] A Cochrane review on treatment for amphetamine, dextroamphetamine, and methamphetamine psychosis states that about 5–15% of users fail to recover completely.[67][126] According to the same review, there is at least one trial that shows antipsychotic medications effectively resolve the symptoms of acute amphetamine psychosis.[67] Psychosis rarely arises from therapeutic use.[52][68][69]
Interactions
- Acidifying Agents: Drugs that acidify the urine, such as ascorbic acid, increase urinary excretion of dextroamphetamine, thus decreasing the half-life of dextroamphetamine in the body.[14][127]
- Alkalinizing Agents: Drugs that alkalinize the urine, such as sodium bicarbonate, decrease urinary excretion of dextroamphetamine, thus increasing the half-life of dextroamphetamine in the body.[14][127]
- Monoamine Oxidase Inhibitors: Concomitant use of MAOIs and central nervous system stimulants such as lisdexamfetamine can cause a hypertensive crisis.[14]
Pharmacology
Mechanism of action
Pharmacodynamics of amphetamine in a dopamine neuron
|

Lisdexamfetamine is an inactive prodrug that is converted in the body to dextroamphetamine, a pharmacologically active compound which is responsible for the drug's activity.[135] After oral ingestion, lisdexamfetamine is broken down by enzymes in red blood cells to form L-lysine, a naturally occurring essential amino acid, and dextroamphetamine.[14] The conversion of lisdexamfetamine to dextroamphetamine is not affected by gastrointestinal pH and is unlikely to be affected by alterations in normal gastrointestinal transit times.[14][136]
The optical isomers of amphetamine, i.e., dextroamphetamine and levoamphetamine, are TAAR1 agonists and vesicular monoamine transporter 2 inhibitors that can enter monoamine neurons;[128][129] this allows them to release monoamine neurotransmitters (dopamine, norepinephrine, and serotonin, among others) from their storage sites in the presynaptic neuron, as well as prevent the reuptake of these neurotransmitters from the synaptic cleft.[128][129]
Lisdexamfetamine was developed with the goal of providing a long duration of effect that is consistent throughout the day, with reduced potential for abuse. The attachment of the amino acid lysine slows down the relative amount of dextroamphetamine available to the blood stream. Because no free dextroamphetamine is present in lisdexamfetamine capsules, dextroamphetamine does not become available through mechanical manipulation, such as crushing or simple extraction. A relatively sophisticated biochemical process is needed to produce dextroamphetamine from lisdexamfetamine.[136] As opposed to Adderall, which contains roughly equal parts of racemic amphetamine and dextroamphetamine salts, lisdexamfetamine is a single-enantiomer dextroamphetamine formula.[135][137] Studies conducted show that lisdexamfetamine dimesylate may have less abuse potential than dextroamphetamine and an abuse profile similar to diethylpropion at dosages that are FDA-approved for treatment of ADHD, but still has a high abuse potential when this dosage is exceeded by over 100%.[136]
Pharmacokinetics
The oral bioavailability of amphetamine varies with gastrointestinal pH;[52] it is well absorbed from the gut, and bioavailability is typically over 75% for dextroamphetamine.[138] Amphetamine is a weak base with a pKa of 9.9;[139] consequently, when the pH is basic, more of the drug is in its lipid soluble free base form, and more is absorbed through the lipid-rich cell membranes of the gut epithelium.[139][52] Conversely, an acidic pH means the drug is predominantly in a water-soluble cationic (salt) form, and less is absorbed.[139] Approximately 15–40% of amphetamine circulating in the bloodstream is bound to plasma proteins.[140] Following absorption, amphetamine readily distributes into most tissues in the body, with high concentrations occurring in cerebrospinal fluid and brain tissue.[141]
The half-lives of amphetamine enantiomers differ and vary with urine pH.[139] At normal urine pH, the half-lives of dextroamphetamine and levoamphetamine are 9–11 hours and 11–14 hours, respectively.[139] Highly acidic urine will reduce the enantiomer half-lives to 7 hours;[141] highly alkaline urine will increase the half-lives up to 34 hours.[141] The immediate-release and extended release variants of salts of both isomers reach peak plasma concentrations at 3 hours and 7 hours post-dose respectively.[139] Amphetamine is eliminated via the kidneys, with 30–40% of the drug being excreted unchanged at normal urinary pH.[139] When the urinary pH is basic, amphetamine is in its free base form, so less is excreted.[139] When urine pH is abnormal, the urinary recovery of amphetamine may range from a low of 1% to a high of 75%, depending mostly upon whether urine is too basic or acidic, respectively.[139] Following oral administration, amphetamine appears in urine within 3 hours.[141] Roughly 90% of ingested amphetamine is eliminated 3 days after the last oral dose.[141]
The prodrug lisdexamfetamine is not as sensitive to pH as amphetamine when being absorbed in the gastrointestinal tract;[14] following absorption into the blood stream, it is converted by red blood cell-associated enzymes to dextroamphetamine via hydrolysis.[14] The elimination half-life of lisdexamfetamine is generally less than 1 hour.[14]
CYP2D6, dopamine β-hydroxylase (DBH), flavin-containing monooxygenase 3 (FMO3), butyrate-CoA ligase (XM-ligase), and glycine N-acyltransferase (GLYAT) are the enzymes known to metabolize amphetamine or its metabolites in humans.[sources 10] Amphetamine has a variety of excreted metabolic products, including 4-hydroxyamphetamine, 4-hydroxynorephedrine, 4-hydroxyphenylacetone, benzoic acid, hippuric acid, norephedrine, and phenylacetone.[139][142] Among these metabolites, the active sympathomimetics are 4-hydroxyamphetamine,[143] 4-hydroxynorephedrine,[144] and norephedrine.[145] The main metabolic pathways involve aromatic para-hydroxylation, aliphatic alpha- and beta-hydroxylation, N-oxidation, N-dealkylation, and deamination.[139][146] The known metabolic pathways, detectable metabolites, and metabolizing enzymes in humans include the following:
Metabolic pathways of amphetamine in humans[sources 10]
|
Chemistry
Lisdexamfetamine is a substituted amphetamine with an amide linkage formed by the condensation of dextroamphetamine with the carboxylate group of the essential amino acid L-lysine.[9] The reaction occurs with retention of stereochemistry, so the product lisdexamfetamine exists as a single stereoisomer. There are many possible names for lisdexamfetamine based on IUPAC nomenclature, but it is usually named as N-[(2S)-1-phenyl-2-propanyl]-L-lysinamide or (2S)-2,6-diamino-N-[(1S)-1-methyl-2-phenylethyl]hexanamide.[156] The condensation reaction occurs with loss of water:
- (S)-PhCH
2CH(CH
3)NH
2 + (S)-HOOCCH(NH
2)CH
2CH
2CH
2CH
2NH
2 → (S,S)-PhCH
2CH(CH
3)NHC(O)CH(NH
2)CH
2CH
2CH
2CH
2NH
2 + H
2O
Amine functional groups are vulnerable to oxidation in air and so pharmaceuticals containing them are usually formulated as salts where this moiety has been protonated. This increases stability, water solubility, and, by converting a molecular compound to an ionic compound, increases the melting point and thereby ensures a solid product.[157] In the case of lisdexamfetamine, this is achieved by reacting with two equivalents of methanesulfonic acid to produce the dimesylate salt, a water-soluble (792 mg mL−1) powder with a white to off-white color.[14]
- PhCH
2CH(CH
3)NHC(O)CH(NH
2)CH
2CH
2CH
2CH
2NH
2 + 2 CH
3SO
3H → [PhCH
2CH(CH
3)NHC(O)CH(NH+
3)CH
2CH
2CH
2CH
2NH+
3][CH
3SO−
3]
2
Comparison to other formulations
Lisdexamfetamine dimesylate is one marketed formulation delivering dextroamphetamine. The following table compares the drug to other amphetamine pharmaceuticals.
| drug | formula | molecular mass [note 9] |
amphetamine base [note 10] |
amphetamine base in equal doses |
doses with equal base content [note 11] | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| (g/mol) | (percent) | (30 mg dose) | ||||||||
| total | base | total | dextro- | levo- | dextro- | levo- | ||||
| dextroamphetamine sulfate[159][160] | (C9H13N)2•H2SO4 | |||||||||
| amphetamine sulfate[161] | (C9H13N)2•H2SO4 | |||||||||
| Adderall | ||||||||||
| 25% | dextroamphetamine sulfate[159][160] | (C9H13N)2•H2SO4 | ||||||||
| 25% | amphetamine sulfate[161] | (C9H13N)2•H2SO4 | ||||||||
| 25% | dextroamphetamine saccharate[162] | (C9H13N)2•C6H10O8 | ||||||||
| 25% | amphetamine aspartate monohydrate[163] | (C9H13N)•C4H7NO4•H2O | ||||||||
| lisdexamfetamine dimesylate[14] | C15H25N3O•(CH4O3S)2 | |||||||||
| amphetamine base suspension[note 12] | C9H13N | |||||||||
History, society, and culture
Lisdexamfetamine was developed by New River Pharmaceuticals, who were bought by Takeda Pharmaceuticals through its acquisition of Shire Pharmaceuticals, shortly before it began being marketed. It was developed with the intention of creating a longer-lasting and less-easily abused version of dextroamphetamine, as the requirement of conversion into dextroamphetamine via enzymes in the red blood cells delays its onset of action, regardless of the route of administration.[164]
On 23 April 2008, the FDA approved lisdexamfetamine for treatment of ADHD in adults.[165] On 19 February 2009, Health Canada approved 30 mg and 50 mg capsules of lisdexamfetamine for treatment of ADHD.[166]
In January 2015, lisdexamfetamine was approved by the U.S. Food and Drug Administration for treatment of binge eating disorder in adults.[167]
Production quotas for 2016 in the United States were 29,750 kilograms.[168]
Names
Lisdexamfetamine is a contraction of L-lysine-dextroamphetamine.
As of July 2014 lisdexamfetamine was sold under the following brands: Elvanse, Samexid, Tyvense, Venvanse, and Vyvanse.[169]
Research
Depression
Some clinical trials that used lisdexamfetamine as an add-on therapy with a selective serotonin reuptake inhibitor (SSRI) or serotonin-norepinephrine reuptake inhibitor (SNRI) for treatment-resistant depression indicated that this is no more effective than the use of an SSRI or SNRI alone.[170] Other studies indicated that psychostimulants potentiated antidepressants, and were under-prescribed for treatment resistant depression. In those studies, patients showed significant improvement in energy, mood, and psychomotor activity.[171] In February 2014, Shire announced that two late-stage clinical trials had shown that Vyvanse was not an effective treatment for depression.[172]
Notes
- The ADHD-related outcome domains with the greatest proportion of significantly improved outcomes from long-term continuous stimulant therapy include academics (≈55% of academic outcomes improved), driving (100% of driving outcomes improved), non-medical drug use (47% of addiction-related outcomes improved), obesity (≈65% of obesity-related outcomes improved), self-esteem (50% of self-esteem outcomes improved), and social function (67% of social function outcomes improved).[22]
The largest effect sizes for outcome improvements from long-term stimulant therapy occur in the domains involving academics (e.g., grade point average, achievement test scores, length of education, and education level), self-esteem (e.g., self-esteem questionnaire assessments, number of suicide attempts, and suicide rates), and social function (e.g., peer nomination scores, social skills, and quality of peer, family, and romantic relationships).[22]
Long-term combination therapy for ADHD (i.e., treatment with both a stimulant and behavioral therapy) produces even larger effect sizes for outcome improvements and improves a larger proportion of outcomes across each domain compared to long-term stimulant therapy alone.[22] - Cochrane reviews are high quality meta-analytic systematic reviews of randomized controlled trials.[29]
- Transcription factors are proteins that increase or decrease the expression of specific genes.[95]
- In simpler terms, this necessary and sufficient relationship means that ΔFosB overexpression in the nucleus accumbens and addiction-related behavioral and neural adaptations always occur together and never occur alone.
- NMDA receptors are voltage-dependent ligand-gated ion channels that requires simultaneous binding of glutamate and a co-agonist (D-serine or glycine) to open the ion channel.[110]
- The review indicated that magnesium L-aspartate and magnesium chloride produce significant changes in addictive behavior;[86] other forms of magnesium were not mentioned.
- The 95% confidence interval indicates that there is a 95% probability that the true number of deaths lies between 3,425 and 4,145.
- 4-Hydroxyamphetamine has been shown to be metabolized into 4-hydroxynorephedrine by dopamine beta-hydroxylase (DBH) in vitro and it is presumed to be metabolized similarly in vivo.[147][151] Evidence from studies that measured the effect of serum DBH concentrations on 4-hydroxyamphetamine metabolism in humans suggests that a different enzyme may mediate the conversion of 4-hydroxyamphetamine to 4-hydroxynorephedrine;[151][153] however, other evidence from animal studies suggests that this reaction is catalyzed by DBH in synaptic vesicles within noradrenergic neurons in the brain.[154][155]
- For uniformity, molecular masses were calculated using the Lenntech Molecular Weight Calculator[158] and were within 0.01g/mol of published pharmaceutical values.
- Amphetamine base percentage = molecular massbase / molecular masstotal. Amphetamine base percentage for Adderall = sum of component percentages / 4.
- dose = (1 / amphetamine base percentage) × scaling factor = (molecular masstotal / molecular massbase) × scaling factor. The values in this column were scaled to a 30 mg dose of dextroamphetamine sulfate. Due to pharmacological differences between these medications (e.g., differences in the release, absorption, conversion, concentration, differing effects of enantiomers, half-life, etc.), the listed values should not be considered equipotent doses.
- This product (Dyanavel XR) is an oral suspension (i.e., a drug that is suspended in a liquid and taken by mouth) that contains 2.5 mg/mL of amphetamine base. The amphetamine base contains dextro- to levo-amphetamine in a ratio of 3.2:1, which is approximately the ratio in Adderall. The product uses an ion exchange resin to achieve extended release of the amphetamine base.
- Image legend
Reference notes
References
- "Lisdexamfetamine Dimesylate Monograph for Professionals". Drugs.com. American Society of Health-System Pharmacists. Retrieved 15 April 2019.
- Stahl SM (March 2017). "Lisdexamfetamine". Prescriber's Guide: Stahl's Essential Psychopharmacology (6th ed.). Cambridge, United Kingdom: Cambridge University Press. pp. 379–384. ISBN 9781108228749.
- "Public Assessment Report Decentralised Procedure" (PDF). MHRA. p. 14. Retrieved 23 August 2014.
- Millichap JG (2010). "Chapter 9: Medications for ADHD". In Millichap JG (ed.). Attention Deficit Hyperactivity Disorder Handbook: A Physician's Guide to ADHD (2nd ed.). New York, USA: Springer. p. 112. ISBN 9781441913968.
Table 9.2 Dextroamphetamine formulations of stimulant medication
Dexedrine [Peak:2–3 h] [Duration:5–6 h] ...
Adderall [Peak:2–3 h] [Duration:5–7 h]
Dexedrine spansules [Peak:7–8 h] [Duration:12 h] ...
Adderall XR [Peak:7–8 h] [Duration:12 h]
Vyvanse [Peak:3–4 h] [Duration:12 h] - Brams M, Mao AR, Doyle RL (September 2008). "Onset of efficacy of long-acting psychostimulants in pediatric attention-deficit/hyperactivity disorder". Postgrad. Med. 120 (3): 69–88. doi:10.3810/pgm.2008.09.1909. PMID 18824827.
Onset of efficacy was earliest for d-MPH-ER at 0.5 hours, followed by d, l-MPH-LA at 1 to 2 hours, MCD at 1.5 hours, d, l-MPH-OR at 1 to 2 hours, MAS-XR at 1.5 to 2 hours, MTS at 2 hours, and LDX at approximately 2 hours. ... MAS-XR, and LDX have a long duration of action at 12 hours postdose
- "Lisdexamfetamine (Vyvanse) Use During Pregnancy". Drugs.com. Retrieved 16 April 2019.
- British national formulary: BNF 76 (76 ed.). Pharmaceutical Press. 2018. pp. 348–349. ISBN 9780857113382.
- Heal DJ, Smith SL, Gosden J, Nutt DJ (June 2013). "Amphetamine, past and present – a pharmacological and clinical perspective". J. Psychopharmacol. 27 (6): 479–496. doi:10.1177/0269881113482532. PMC 3666194. PMID 23539642.
- Blick SK, Keating GM (2007). "Lisdexamfetamine". Paediatric Drugs. 9 (2): 129–135, discussion 136–138. doi:10.2165/00148581-200709020-00007. PMID 17407369.
- "NADAC as of 2019-02-27". Centers for Medicare and Medicaid Services. Retrieved 3 March 2019.
- "The Top 300 of 2020". ClinCalc. Retrieved 11 April 2020.
- "Lisdexamfetamine Dimesylate - Drug Usage Statistics". ClinCalc. Retrieved 11 April 2020.
- Drugs of Abuse (PDF). Drug Enforcement Administration • U.S. Department of Justice. 2017. p. 22. Retrieved 16 April 2019.
- "Vyvanse- lisdexamfetamine dimesylate capsule Vyvanse- lisdexamfetamine dimesylate tablet, chewable". DailyMed. Shire US Inc. 30 October 2019. Retrieved 22 December 2019.
- Carvalho M, Carmo H, Costa VM, Capela JP, Pontes H, Remião F, Carvalho F, Bastos Mde L (August 2012). "Toxicity of amphetamines: an update". Archives of Toxicology. 86 (8): 1167–1231. doi:10.1007/s00204-012-0815-5. PMID 22392347.
- Berman S, O'Neill J, Fears S, Bartzokis G, London ED (October 2008). "Abuse of amphetamines and structural abnormalities in the brain". Annals of the New York Academy of Sciences. 1141 (1): 195–220. doi:10.1196/annals.1441.031. PMC 2769923. PMID 18991959.
- Hart H, Radua J, Nakao T, Mataix-Cols D, Rubia K (February 2013). "Meta-analysis of functional magnetic resonance imaging studies of inhibition and attention in attention-deficit/hyperactivity disorder: exploring task-specific, stimulant medication, and age effects". JAMA Psychiatry. 70 (2): 185–198. doi:10.1001/jamapsychiatry.2013.277. PMID 23247506.
- Spencer TJ, Brown A, Seidman LJ, Valera EM, Makris N, Lomedico A, Faraone SV, Biederman J (September 2013). "Effect of psychostimulants on brain structure and function in ADHD: a qualitative literature review of magnetic resonance imaging-based neuroimaging studies". The Journal of Clinical Psychiatry. 74 (9): 902–917. doi:10.4088/JCP.12r08287. PMC 3801446. PMID 24107764.
- Frodl T, Skokauskas N (February 2012). "Meta-analysis of structural MRI studies in children and adults with attention deficit hyperactivity disorder indicates treatment effects". Acta Psychiatrica Scandinavica. 125 (2): 114–126. doi:10.1111/j.1600-0447.2011.01786.x. PMID 22118249.
Basal ganglia regions like the right globus pallidus, the right putamen, and the nucleus caudatus are structurally affected in children with ADHD. These changes and alterations in limbic regions like ACC and amygdala are more pronounced in non-treated populations and seem to diminish over time from child to adulthood. Treatment seems to have positive effects on brain structure.
- Huang YS, Tsai MH (July 2011). "Long-term outcomes with medications for attention-deficit hyperactivity disorder: current status of knowledge". CNS Drugs. 25 (7): 539–554. doi:10.2165/11589380-000000000-00000. PMID 21699268.
Several other studies,[97-101] including a meta-analytic review[98] and a retrospective study,[97] suggested that stimulant therapy in childhood is associated with a reduced risk of subsequent substance use, cigarette smoking and alcohol use disorders. ... Recent studies have demonstrated that stimulants, along with the non-stimulants atomoxetine and extended-release guanfacine, are continuously effective for more than 2-year treatment periods with few and tolerable adverse effects. The effectiveness of long-term therapy includes not only the core symptoms of ADHD, but also improved quality of life and academic achievements. The most concerning short-term adverse effects of stimulants, such as elevated blood pressure and heart rate, waned in long-term follow-up studies. ... The current data do not support the potential impact of stimulants on the worsening or development of tics or substance abuse into adulthood. In the longest follow-up study (of more than 10 years), lifetime stimulant treatment for ADHD was effective and protective against the development of adverse psychiatric disorders.
- Millichap JG (2010). "Chapter 9: Medications for ADHD". In Millichap JG (ed.). Attention Deficit Hyperactivity Disorder Handbook: A Physician's Guide to ADHD (2nd ed.). New York, USA: Springer. pp. 121–123, 125–127. ISBN 9781441913968.
Ongoing research has provided answers to many of the parents' concerns, and has confirmed the effectiveness and safety of the long-term use of medication.
- Arnold LE, Hodgkins P, Caci H, Kahle J, Young S (February 2015). "Effect of treatment modality on long-term outcomes in attention-deficit/hyperactivity disorder: a systematic review". PLOS ONE. 10 (2): e0116407. doi:10.1371/journal.pone.0116407. PMC 4340791. PMID 25714373.
The highest proportion of improved outcomes was reported with combination treatment (83% of outcomes). Among significantly improved outcomes, the largest effect sizes were found for combination treatment. The greatest improvements were associated with academic, self-esteem, or social function outcomes.
Figure 3: Treatment benefit by treatment type and outcome group - Malenka RC, Nestler EJ, Hyman SE (2009). "Chapter 6: Widely Projecting Systems: Monoamines, Acetylcholine, and Orexin". In Sydor A, Brown RY (eds.). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York, USA: McGraw-Hill Medical. pp. 154–157. ISBN 9780071481274.
- Malenka RC, Nestler EJ, Hyman SE (2009). "Chapter 13: Higher Cognitive Function and Behavioral Control". In Sydor A, Brown RY (eds.). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York, USA: McGraw-Hill Medical. pp. 318, 321. ISBN 9780071481274.
Therapeutic (relatively low) doses of psychostimulants, such as methylphenidate and amphetamine, improve performance on working memory tasks both in normal subjects and those with ADHD. ... stimulants act not only on working memory function, but also on general levels of arousal and, within the nucleus accumbens, improve the saliency of tasks. Thus, stimulants improve performance on effortful but tedious tasks ... through indirect stimulation of dopamine and norepinephrine receptors. ...
Beyond these general permissive effects, dopamine (acting via D1 receptors) and norepinephrine (acting at several receptors) can, at optimal levels, enhance working memory and aspects of attention. - Bidwell LC, McClernon FJ, Kollins SH (August 2011). "Cognitive enhancers for the treatment of ADHD". Pharmacology Biochemistry and Behavior. 99 (2): 262–274. doi:10.1016/j.pbb.2011.05.002. PMC 3353150. PMID 21596055.
- Parker J, Wales G, Chalhoub N, Harpin V (September 2013). "The long-term outcomes of interventions for the management of attention-deficit hyperactivity disorder in children and adolescents: a systematic review of randomized controlled trials". Psychology Research and Behavior Management. 6: 87–99. doi:10.2147/PRBM.S49114. PMC 3785407. PMID 24082796.
Only one paper53 examining outcomes beyond 36 months met the review criteria. ... There is high level evidence suggesting that pharmacological treatment can have a major beneficial effect on the core symptoms of ADHD (hyperactivity, inattention, and impulsivity) in approximately 80% of cases compared with placebo controls, in the short term.
- Millichap JG (2010). "Chapter 9: Medications for ADHD". In Millichap JG (ed.). Attention Deficit Hyperactivity Disorder Handbook: A Physician's Guide to ADHD (2nd ed.). New York, USA: Springer. pp. 111–113. ISBN 9781441913968.
- "Stimulants for Attention Deficit Hyperactivity Disorder". WebMD. Healthwise. 12 April 2010. Retrieved 12 November 2013.
- Scholten RJ, Clarke M, Hetherington J (August 2005). "The Cochrane Collaboration". European Journal of Clinical Nutrition. 59 (Suppl 1): S147–S149, discussion S195–S196. doi:10.1038/sj.ejcn.1602188. PMID 16052183.
- Castells X, Blanco-Silvente L, Cunill R (August 2018). "Amphetamines for attention deficit hyperactivity disorder (ADHD) in adults". Cochrane Database of Systematic Reviews. 8: CD007813. doi:10.1002/14651858.CD007813.pub3. PMC 6513464. PMID 30091808.
- Punja S, Shamseer L, Hartling L, Urichuk L, Vandermeer B, Nikles J, Vohra S (February 2016). "Amphetamines for attention deficit hyperactivity disorder (ADHD) in children and adolescents". Cochrane Database of Systematic Reviews. 2: CD009996. doi:10.1002/14651858.CD009996.pub2. PMID 26844979.
- Osland ST, Steeves TD, Pringsheim T (June 2018). "Pharmacological treatment for attention deficit hyperactivity disorder (ADHD) in children with comorbid tic disorders". Cochrane Database of Systematic Reviews. 6: CD007990. doi:10.1002/14651858.CD007990.pub3. PMC 6513283. PMID 29944175.
- Spencer RC, Devilbiss DM, Berridge CW (June 2015). "The Cognition-Enhancing Effects of Psychostimulants Involve Direct Action in the Prefrontal Cortex". Biological Psychiatry. 77 (11): 940–950. doi:10.1016/j.biopsych.2014.09.013. PMC 4377121. PMID 25499957.
The procognitive actions of psychostimulants are only associated with low doses. Surprisingly, despite nearly 80 years of clinical use, the neurobiology of the procognitive actions of psychostimulants has only recently been systematically investigated. Findings from this research unambiguously demonstrate that the cognition-enhancing effects of psychostimulants involve the preferential elevation of catecholamines in the PFC and the subsequent activation of norepinephrine α2 and dopamine D1 receptors. ... This differential modulation of PFC-dependent processes across dose appears to be associated with the differential involvement of noradrenergic α2 versus α1 receptors. Collectively, this evidence indicates that at low, clinically relevant doses, psychostimulants are devoid of the behavioral and neurochemical actions that define this class of drugs and instead act largely as cognitive enhancers (improving PFC-dependent function). ... In particular, in both animals and humans, lower doses maximally improve performance in tests of working memory and response inhibition, whereas maximal suppression of overt behavior and facilitation of attentional processes occurs at higher doses.
- Ilieva IP, Hook CJ, Farah MJ (June 2015). "Prescription Stimulants' Effects on Healthy Inhibitory Control, Working Memory, and Episodic Memory: A Meta-analysis". Journal of Cognitive Neuroscience. 27 (6): 1069–1089. doi:10.1162/jocn_a_00776. PMID 25591060.
Specifically, in a set of experiments limited to high-quality designs, we found significant enhancement of several cognitive abilities. ... The results of this meta-analysis ... do confirm the reality of cognitive enhancing effects for normal healthy adults in general, while also indicating that these effects are modest in size.
- Bagot KS, Kaminer Y (April 2014). "Efficacy of stimulants for cognitive enhancement in non-attention deficit hyperactivity disorder youth: a systematic review". Addiction. 109 (4): 547–557. doi:10.1111/add.12460. PMC 4471173. PMID 24749160.
Amphetamine has been shown to improve consolidation of information (0.02 ≥ P ≤ 0.05), leading to improved recall.
- Devous MD, Trivedi MH, Rush AJ (April 2001). "Regional cerebral blood flow response to oral amphetamine challenge in healthy volunteers". Journal of Nuclear Medicine. 42 (4): 535–542. PMID 11337538.
- Malenka RC, Nestler EJ, Hyman SE (2009). "Chapter 10: Neural and Neuroendocrine Control of the Internal Milieu". In Sydor A, Brown RY (eds.). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York, USA: McGraw-Hill Medical. p. 266. ISBN 9780071481274.
Dopamine acts in the nucleus accumbens to attach motivational significance to stimuli associated with reward.
- Wood S, Sage JR, Shuman T, Anagnostaras SG (January 2014). "Psychostimulants and cognition: a continuum of behavioral and cognitive activation". Pharmacological Reviews. 66 (1): 193–221. doi:10.1124/pr.112.007054. PMC 3880463. PMID 24344115.
- Twohey M (26 March 2006). "Pills become an addictive study aid". JS Online. Archived from the original on 15 August 2007. Retrieved 2 December 2007.
- Teter CJ, McCabe SE, LaGrange K, Cranford JA, Boyd CJ (October 2006). "Illicit use of specific prescription stimulants among college students: prevalence, motives, and routes of administration". Pharmacotherapy. 26 (10): 1501–1510. doi:10.1592/phco.26.10.1501. PMC 1794223. PMID 16999660.
- Weyandt LL, Oster DR, Marraccini ME, Gudmundsdottir BG, Munro BA, Zavras BM, Kuhar B (September 2014). "Pharmacological interventions for adolescents and adults with ADHD: stimulant and nonstimulant medications and misuse of prescription stimulants". Psychology Research and Behavior Management. 7: 223–249. doi:10.2147/PRBM.S47013. PMC 4164338. PMID 25228824.
misuse of prescription stimulants has become a serious problem on college campuses across the US and has been recently documented in other countries as well. ... Indeed, large numbers of students claim to have engaged in the nonmedical use of prescription stimulants, which is reflected in lifetime prevalence rates of prescription stimulant misuse ranging from 5% to nearly 34% of students.
- Clemow DB, Walker DJ (September 2014). "The potential for misuse and abuse of medications in ADHD: a review". Postgraduate Medicine. 126 (5): 64–81. doi:10.3810/pgm.2014.09.2801. PMID 25295651.
Overall, the data suggest that ADHD medication misuse and diversion are common health care problems for stimulant medications, with the prevalence believed to be approximately 5% to 10% of high school students and 5% to 35% of college students, depending on the study.
- Liddle DG, Connor DJ (June 2013). "Nutritional supplements and ergogenic AIDS". Primary Care: Clinics in Office Practice. 40 (2): 487–505. doi:10.1016/j.pop.2013.02.009. PMID 23668655.
Amphetamines and caffeine are stimulants that increase alertness, improve focus, decrease reaction time, and delay fatigue, allowing for an increased intensity and duration of training ...
Physiologic and performance effects
• Amphetamines increase dopamine/norepinephrine release and inhibit their reuptake, leading to central nervous system (CNS) stimulation
• Amphetamines seem to enhance athletic performance in anaerobic conditions 39 40
• Improved reaction time
• Increased muscle strength and delayed muscle fatigue
• Increased acceleration
• Increased alertness and attention to task - Westfall DP, Westfall TC (2010). "Miscellaneous Sympathomimetic Agonists". In Brunton LL, Chabner BA, Knollmann BC (eds.). Goodman & Gilman's Pharmacological Basis of Therapeutics (12th ed.). New York, USA: McGraw-Hill. ISBN 9780071624428.
- Bracken NM (January 2012). "National Study of Substance Use Trends Among NCAA College Student-Athletes" (PDF). NCAA Publications. National Collegiate Athletic Association. Retrieved 8 October 2013.
- Docherty JR (June 2008). "Pharmacology of stimulants prohibited by the World Anti-Doping Agency (WADA)". British Journal of Pharmacology. 154 (3): 606–622. doi:10.1038/bjp.2008.124. PMC 2439527. PMID 18500382.
- Parr JW (July 2011). "Attention-deficit hyperactivity disorder and the athlete: new advances and understanding". Clinics in Sports Medicine. 30 (3): 591–610. doi:10.1016/j.csm.2011.03.007. PMID 21658550.
In 1980, Chandler and Blair47 showed significant increases in knee extension strength, acceleration, anaerobic capacity, time to exhaustion during exercise, pre-exercise and maximum heart rates, and time to exhaustion during maximal oxygen consumption (VO2 max) testing after administration of 15 mg of dextroamphetamine versus placebo. Most of the information to answer this question has been obtained in the past decade through studies of fatigue rather than an attempt to systematically investigate the effect of ADHD drugs on exercise.
- Roelands B, de Koning J, Foster C, Hettinga F, Meeusen R (May 2013). "Neurophysiological determinants of theoretical concepts and mechanisms involved in pacing". Sports Medicine. 43 (5): 301–311. doi:10.1007/s40279-013-0030-4. PMID 23456493.
In high-ambient temperatures, dopaminergic manipulations clearly improve performance. The distribution of the power output reveals that after dopamine reuptake inhibition, subjects are able to maintain a higher power output compared with placebo. ... Dopaminergic drugs appear to override a safety switch and allow athletes to use a reserve capacity that is 'off-limits' in a normal (placebo) situation.
- Parker KL, Lamichhane D, Caetano MS, Narayanan NS (October 2013). "Executive dysfunction in Parkinson's disease and timing deficits". Frontiers in Integrative Neuroscience. 7: 75. doi:10.3389/fnint.2013.00075. PMC 3813949. PMID 24198770.
Manipulations of dopaminergic signaling profoundly influence interval timing, leading to the hypothesis that dopamine influences internal pacemaker, or "clock," activity. For instance, amphetamine, which increases concentrations of dopamine at the synaptic cleft advances the start of responding during interval timing, whereas antagonists of D2 type dopamine receptors typically slow timing;... Depletion of dopamine in healthy volunteers impairs timing, while amphetamine releases synaptic dopamine and speeds up timing.
- Rattray B, Argus C, Martin K, Northey J, Driller M (March 2015). "Is it time to turn our attention toward central mechanisms for post-exertional recovery strategies and performance?". Frontiers in Physiology. 6: 79. doi:10.3389/fphys.2015.00079. PMC 4362407. PMID 25852568.
Aside from accounting for the reduced performance of mentally fatigued participants, this model rationalizes the reduced RPE and hence improved cycling time trial performance of athletes using a glucose mouthwash (Chambers et al., 2009) and the greater power output during a RPE matched cycling time trial following amphetamine ingestion (Swart, 2009). ... Dopamine stimulating drugs are known to enhance aspects of exercise performance (Roelands et al., 2008)
- Roelands B, De Pauw K, Meeusen R (June 2015). "Neurophysiological effects of exercise in the heat". Scandinavian Journal of Medicine & Science in Sports. 25 (Suppl 1): 65–78. doi:10.1111/sms.12350. PMID 25943657.
This indicates that subjects did not feel they were producing more power and consequently more heat. The authors concluded that the "safety switch" or the mechanisms existing in the body to prevent harmful effects are overridden by the drug administration (Roelands et al., 2008b). Taken together, these data indicate strong ergogenic effects of an increased DA concentration in the brain, without any change in the perception of effort.
- "Adderall XR- dextroamphetamine sulfate, dextroamphetamine saccharate, amphetamine sulfate and amphetamine aspartate capsule, extended release". DailyMed. Shire US Inc. 17 July 2019. Retrieved 22 December 2019.
- Heedes G, Ailakis J. "Amphetamine (PIM 934)". INCHEM. International Programme on Chemical Safety. Retrieved 24 June 2014.
- "Adderall XR Prescribing Information" (PDF). United States Food and Drug Administration. Shire US Inc. December 2013. pp. 4–6. Retrieved 30 December 2013.
- "FDA Pregnancy Categories" (PDF). United States Food and Drug Administration. 21 October 2004. Archived from the original (PDF) on 14 December 2010. Retrieved 31 October 2013.
- "Dexamphetamine tablets". Therapeutic Goods Administration. Retrieved 12 April 2014.
- Vitiello B (April 2008). "Understanding the risk of using medications for attention deficit hyperactivity disorder with respect to physical growth and cardiovascular function". Child and Adolescent Psychiatric Clinics of North America. 17 (2): 459–474. doi:10.1016/j.chc.2007.11.010. PMC 2408826. PMID 18295156.
- Stahl SM (March 2017). "Amphetamine (D,L)". Prescriber's Guide: Stahl's Essential Psychopharmacology (6th ed.). Cambridge, United Kingdom: Cambridge University Press. pp. 45–51. ISBN 9781108228749. Retrieved 5 August 2017.
- Ramey JT, Bailen E, Lockey RF (2006). "Rhinitis medicamentosa" (PDF). Journal of Investigational Allergology & Clinical Immunology. 16 (3): 148–155. PMID 16784007. Retrieved 29 April 2015.
Table 2. Decongestants Causing Rhinitis Medicamentosa
– Nasal decongestants:
– Sympathomimetic:
• Amphetamine - "FDA Drug Safety Communication: Safety Review Update of Medications used to treat Attention-Deficit/Hyperactivity Disorder (ADHD) in children and young adults". United States Food and Drug Administration. 1 November 2011. Archived from the original on 25 August 2019. Retrieved 24 December 2019.
- Cooper WO, Habel LA, Sox CM, Chan KA, Arbogast PG, Cheetham TC, Murray KT, Quinn VP, Stein CM, Callahan ST, Fireman BH, Fish FA, Kirshner HS, O'Duffy A, Connell FA, Ray WA (November 2011). "ADHD drugs and serious cardiovascular events in children and young adults". New England Journal of Medicine. 365 (20): 1896–1904. doi:10.1056/NEJMoa1110212. PMC 4943074. PMID 22043968.
- "FDA Drug Safety Communication: Safety Review Update of Medications used to treat Attention-Deficit/Hyperactivity Disorder (ADHD) in adults". United States Food and Drug Administration. 12 December 2011. Archived from the original on 14 December 2019. Retrieved 24 December 2013.
- Habel LA, Cooper WO, Sox CM, Chan KA, Fireman BH, Arbogast PG, Cheetham TC, Quinn VP, Dublin S, Boudreau DM, Andrade SE, Pawloski PA, Raebel MA, Smith DH, Achacoso N, Uratsu C, Go AS, Sidney S, Nguyen-Huynh MN, Ray WA, Selby JV (December 2011). "ADHD medications and risk of serious cardiovascular events in young and middle-aged adults". JAMA. 306 (24): 2673–2683. doi:10.1001/jama.2011.1830. PMC 3350308. PMID 22161946.
- Montgomery KA (June 2008). "Sexual desire disorders". Psychiatry. 5 (6): 50–55. PMC 2695750. PMID 19727285.
- O'Connor PG (February 2012). "Amphetamines". Merck Manual for Health Care Professionals. Merck. Retrieved 8 May 2012.
- Shoptaw SJ, Kao U, Ling W (January 2009). Shoptaw SJ, Ali R (ed.). "Treatment for amphetamine psychosis". Cochrane Database of Systematic Reviews (1): CD003026. doi:10.1002/14651858.CD003026.pub3. PMC 7004251. PMID 19160215.
A minority of individuals who use amphetamines develop full-blown psychosis requiring care at emergency departments or psychiatric hospitals. In such cases, symptoms of amphetamine psychosis commonly include paranoid and persecutory delusions as well as auditory and visual hallucinations in the presence of extreme agitation. More common (about 18%) is for frequent amphetamine users to report psychotic symptoms that are sub-clinical and that do not require high-intensity intervention ...
About 5–15% of the users who develop an amphetamine psychosis fail to recover completely (Hofmann 1983) ...
Findings from one trial indicate use of antipsychotic medications effectively resolves symptoms of acute amphetamine psychosis.
psychotic symptoms of individuals with amphetamine psychosis may be due exclusively to heavy use of the drug or heavy use of the drug may exacerbate an underlying vulnerability to schizophrenia. - Bramness JG, Gundersen ØH, Guterstam J, Rognli EB, Konstenius M, Løberg EM, Medhus S, Tanum L, Franck J (December 2012). "Amphetamine-induced psychosis—a separate diagnostic entity or primary psychosis triggered in the vulnerable?". BMC Psychiatry. 12: 221. doi:10.1186/1471-244X-12-221. PMC 3554477. PMID 23216941.
In these studies, amphetamine was given in consecutively higher doses until psychosis was precipitated, often after 100–300 mg of amphetamine ... Secondly, psychosis has been viewed as an adverse event, although rare, in children with ADHD who have been treated with amphetamine
- Greydanus D. "Stimulant Misuse: Strategies to Manage a Growing Problem" (PDF). American College Health Association (Review Article). ACHA Professional Development Program. p. 20. Archived from the original (PDF) on 3 November 2013. Retrieved 2 November 2013.
- Childs E, de Wit H (May 2009). "Amphetamine-induced place preference in humans". Biological Psychiatry. 65 (10): 900–904. doi:10.1016/j.biopsych.2008.11.016. PMC 2693956. PMID 19111278.
This study demonstrates that humans, like nonhumans, prefer a place associated with amphetamine administration. These findings support the idea that subjective responses to a drug contribute to its ability to establish place conditioning.
- Malenka RC, Nestler EJ, Hyman SE (2009). "Chapter 15: Reinforcement and Addictive Disorders". In Sydor A, Brown RY (eds.). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York: McGraw-Hill Medical. pp. 364–375. ISBN 9780071481274.
- Nestler EJ (December 2013). "Cellular basis of memory for addiction". Dialogues in Clinical Neuroscience. 15 (4): 431–443. PMC 3898681. PMID 24459410.
Despite the importance of numerous psychosocial factors, at its core, drug addiction involves a biological process: the ability of repeated exposure to a drug of abuse to induce changes in a vulnerable brain that drive the compulsive seeking and taking of drugs, and loss of control over drug use, that define a state of addiction. ... A large body of literature has demonstrated that such ΔFosB induction in D1-type [nucleus accumbens] neurons increases an animal's sensitivity to drug as well as natural rewards and promotes drug self-administration, presumably through a process of positive reinforcement ... Another ΔFosB target is cFos: as ΔFosB accumulates with repeated drug exposure it represses c-Fos and contributes to the molecular switch whereby ΔFosB is selectively induced in the chronic drug-treated state.41. ... Moreover, there is increasing evidence that, despite a range of genetic risks for addiction across the population, exposure to sufficiently high doses of a drug for long periods of time can transform someone who has relatively lower genetic loading into an addict.
- "Glossary of Terms". Mount Sinai School of Medicine. Department of Neuroscience. Retrieved 9 February 2015.
- Volkow ND, Koob GF, McLellan AT (January 2016). "Neurobiologic Advances from the Brain Disease Model of Addiction". New England Journal of Medicine. 374 (4): 363–371. doi:10.1056/NEJMra1511480. PMC 6135257. PMID 26816013.
Substance-use disorder: A diagnostic term in the fifth edition of the Diagnostic and Statistical Manual of Mental Disorders (DSM-5) referring to recurrent use of alcohol or other drugs that causes clinically and functionally significant impairment, such as health problems, disability, and failure to meet major responsibilities at work, school, or home. Depending on the level of severity, this disorder is classified as mild, moderate, or severe.
Addiction: A term used to indicate the most severe, chronic stage of substance-use disorder, in which there is a substantial loss of self-control, as indicated by compulsive drug taking despite the desire to stop taking the drug. In the DSM-5, the term addiction is synonymous with the classification of severe substance-use disorder. - Renthal W, Nestler EJ (September 2009). "Chromatin regulation in drug addiction and depression". Dialogues in Clinical Neuroscience. 11 (3): 257–268. PMC 2834246. PMID 19877494.
[Psychostimulants] increase cAMP levels in striatum, which activates protein kinase A (PKA) and leads to phosphorylation of its targets. This includes the cAMP response element binding protein (CREB), the phosphorylation of which induces its association with the histone acetyltransferase, CREB binding protein (CBP) to acetylate histones and facilitate gene activation. This is known to occur on many genes including fosB and c-fos in response to psychostimulant exposure. ΔFosB is also upregulated by chronic psychostimulant treatments, and is known to activate certain genes (eg, cdk5) and repress others (eg, c-fos) where it recruits HDAC1 as a corepressor. ... Chronic exposure to psychostimulants increases glutamatergic [signaling] from the prefrontal cortex to the NAc. Glutamatergic signaling elevates Ca2+ levels in NAc postsynaptic elements where it activates CaMK (calcium/calmodulin protein kinases) signaling, which, in addition to phosphorylating CREB, also phosphorylates HDAC5.
Figure 2: Psychostimulant-induced signaling events - Broussard JI (January 2012). "Co-transmission of dopamine and glutamate". The Journal of General Physiology. 139 (1): 93–96. doi:10.1085/jgp.201110659. PMC 3250102. PMID 22200950.
Coincident and convergent input often induces plasticity on a postsynaptic neuron. The NAc integrates processed information about the environment from basolateral amygdala, hippocampus, and prefrontal cortex (PFC), as well as projections from midbrain dopamine neurons. Previous studies have demonstrated how dopamine modulates this integrative process. For example, high frequency stimulation potentiates hippocampal inputs to the NAc while simultaneously depressing PFC synapses (Goto and Grace, 2005). The converse was also shown to be true; stimulation at PFC potentiates PFC–NAc synapses but depresses hippocampal–NAc synapses. In light of the new functional evidence of midbrain dopamine/glutamate co-transmission (references above), new experiments of NAc function will have to test whether midbrain glutamatergic inputs bias or filter either limbic or cortical inputs to guide goal-directed behavior.
- Kanehisa Laboratories (10 October 2014). "Amphetamine – Homo sapiens (human)". KEGG Pathway. Retrieved 31 October 2014.
Most addictive drugs increase extracellular concentrations of dopamine (DA) in nucleus accumbens (NAc) and medial prefrontal cortex (mPFC), projection areas of mesocorticolimbic DA neurons and key components of the "brain reward circuit". Amphetamine achieves this elevation in extracellular levels of DA by promoting efflux from synaptic terminals. ... Chronic exposure to amphetamine induces a unique transcription factor delta FosB, which plays an essential role in long-term adaptive changes in the brain.
- Cadet JL, Brannock C, Jayanthi S, Krasnova IN (2015). "Transcriptional and epigenetic substrates of methamphetamine addiction and withdrawal: evidence from a long-access self-administration model in the rat". Molecular Neurobiology. 51 (2): 696–717. doi:10.1007/s12035-014-8776-8. PMC 4359351. PMID 24939695.
Figure 1
- Robison AJ, Nestler EJ (November 2011). "Transcriptional and epigenetic mechanisms of addiction". Nature Reviews Neuroscience. 12 (11): 623–637. doi:10.1038/nrn3111. PMC 3272277. PMID 21989194.
ΔFosB serves as one of the master control proteins governing this structural plasticity. ... ΔFosB also represses G9a expression, leading to reduced repressive histone methylation at the cdk5 gene. The net result is gene activation and increased CDK5 expression. ... In contrast, ΔFosB binds to the c-fos gene and recruits several co-repressors, including HDAC1 (histone deacetylase 1) and SIRT 1 (sirtuin 1). ... The net result is c-fos gene repression.
Figure 4: Epigenetic basis of drug regulation of gene expression - Nestler EJ (December 2012). "Transcriptional mechanisms of drug addiction". Clinical Psychopharmacology and Neuroscience. 10 (3): 136–143. doi:10.9758/cpn.2012.10.3.136. PMC 3569166. PMID 23430970.
The 35-37 kD ΔFosB isoforms accumulate with chronic drug exposure due to their extraordinarily long half-lives. ... As a result of its stability, the ΔFosB protein persists in neurons for at least several weeks after cessation of drug exposure. ... ΔFosB overexpression in nucleus accumbens induces NFκB ... In contrast, the ability of ΔFosB to repress the c-Fos gene occurs in concert with the recruitment of a histone deacetylase and presumably several other repressive proteins such as a repressive histone methyltransferase
- Nestler EJ (October 2008). "Transcriptional mechanisms of addiction: Role of ΔFosB". Philosophical Transactions of the Royal Society B: Biological Sciences. 363 (1507): 3245–3255. doi:10.1098/rstb.2008.0067. PMC 2607320. PMID 18640924.
Recent evidence has shown that ΔFosB also represses the c-fos gene that helps create the molecular switch—from the induction of several short-lived Fos family proteins after acute drug exposure to the predominant accumulation of ΔFosB after chronic drug exposure
- Malenka RC, Nestler EJ, Hyman SE, Holtzman DM (2015). "Chapter 16: Reinforcement and Addictive Disorders". Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (3rd ed.). New York: McGraw-Hill Medical. ISBN 9780071827706.
Such agents also have important therapeutic uses; cocaine, for example, is used as a local anesthetic (Chapter 2), and amphetamines and methylphenidate are used in low doses to treat attention deficit hyperactivity disorder and in higher doses to treat narcolepsy (Chapter 12). Despite their clinical uses, these drugs are strongly reinforcing, and their long-term use at high doses is linked with potential addiction, especially when they are rapidly administered or when high-potency forms are given.
- Kollins SH (May 2008). "A qualitative review of issues arising in the use of psycho-stimulant medications in patients with ADHD and co-morbid substance use disorders". Current Medical Research and Opinion. 24 (5): 1345–1357. doi:10.1185/030079908X280707. PMID 18384709.
When oral formulations of psychostimulants are used at recommended doses and frequencies, they are unlikely to yield effects consistent with abuse potential in patients with ADHD.
- Coghill DR, Caballero B, Sorooshian S, Civil R (June 2014). "A systematic review of the safety of lisdexamfetamine dimesylate". CNS Drugs. 28 (6): 497–511. doi:10.1007/s40263-014-0166-2. PMC 4057639. PMID 24788672.
The prodrug formulation of LDX may also lead to reduced abuse potential of LDX compared with immediate-release d-AMP.
- Kanehisa Laboratories (10 October 2014). "Amphetamine – Homo sapiens (human)". KEGG Pathway. Retrieved 31 October 2014.
- Nechifor M (March 2008). "Magnesium in drug dependences". Magnesium Research. 21 (1): 5–15. doi:10.1684/mrh.2008.0124 (inactive 3 June 2020). PMID 18557129.
- Ruffle JK (November 2014). "Molecular neurobiology of addiction: what's all the (Δ)FosB about?". The American Journal of Drug and Alcohol Abuse. 40 (6): 428–437. doi:10.3109/00952990.2014.933840. PMID 25083822.
ΔFosB is an essential transcription factor implicated in the molecular and behavioral pathways of addiction following repeated drug exposure.
- Robison AJ, Nestler EJ (November 2011). "Transcriptional and epigenetic mechanisms of addiction". Nature Reviews Neuroscience. 12 (11): 623–637. doi:10.1038/nrn3111. PMC 3272277. PMID 21989194.
ΔFosB has been linked directly to several addiction-related behaviors ... Importantly, genetic or viral overexpression of ΔJunD, a dominant negative mutant of JunD which antagonizes ΔFosB- and other AP-1-mediated transcriptional activity, in the NAc or OFC blocks these key effects of drug exposure14,22–24. This indicates that ΔFosB is both necessary and sufficient for many of the changes wrought in the brain by chronic drug exposure. ΔFosB is also induced in D1-type NAc MSNs by chronic consumption of several natural rewards, including sucrose, high fat food, sex, wheel running, where it promotes that consumption14,26–30. This implicates ΔFosB in the regulation of natural rewards under normal conditions and perhaps during pathological addictive-like states. ... ΔFosB serves as one of the master control proteins governing this structural plasticity.
- Olsen CM (December 2011). "Natural rewards, neuroplasticity, and non-drug addictions". Neuropharmacology. 61 (7): 1109–1122. doi:10.1016/j.neuropharm.2011.03.010. PMC 3139704. PMID 21459101.
Similar to environmental enrichment, studies have found that exercise reduces self-administration and relapse to drugs of abuse (Cosgrove et al., 2002; Zlebnik et al., 2010). There is also some evidence that these preclinical findings translate to human populations, as exercise reduces withdrawal symptoms and relapse in abstinent smokers (Daniel et al., 2006; Prochaska et al., 2008), and one drug recovery program has seen success in participants that train for and compete in a marathon as part of the program (Butler, 2005). ... In humans, the role of dopamine signaling in incentive-sensitization processes has recently been highlighted by the observation of a dopamine dysregulation syndrome in some patients taking dopaminergic drugs. This syndrome is characterized by a medication-induced increase in (or compulsive) engagement in non-drug rewards such as gambling, shopping, or sex (Evans et al., 2006; Aiken, 2007; Lader, 2008).
- Lynch WJ, Peterson AB, Sanchez V, Abel J, Smith MA (September 2013). "Exercise as a novel treatment for drug addiction: a neurobiological and stage-dependent hypothesis". Neuroscience & Biobehavioral Reviews. 37 (8): 1622–1644. doi:10.1016/j.neubiorev.2013.06.011. PMC 3788047. PMID 23806439.
These findings suggest that exercise may "magnitude"-dependently prevent the development of an addicted phenotype possibly by blocking/reversing behavioral and neuroadaptive changes that develop during and following extended access to the drug. ... Exercise has been proposed as a treatment for drug addiction that may reduce drug craving and risk of relapse. Although few clinical studies have investigated the efficacy of exercise for preventing relapse, the few studies that have been conducted generally report a reduction in drug craving and better treatment outcomes ... Taken together, these data suggest that the potential benefits of exercise during relapse, particularly for relapse to psychostimulants, may be mediated via chromatin remodeling and possibly lead to greater treatment outcomes.
- Zhou Y, Zhao M, Zhou C, Li R (July 2015). "Sex differences in drug addiction and response to exercise intervention: From human to animal studies". Frontiers in Neuroendocrinology. 40: 24–41. doi:10.1016/j.yfrne.2015.07.001. PMC 4712120. PMID 26182835.
Collectively, these findings demonstrate that exercise may serve as a substitute or competition for drug abuse by changing ΔFosB or cFos immunoreactivity in the reward system to protect against later or previous drug use. ... The postulate that exercise serves as an ideal intervention for drug addiction has been widely recognized and used in human and animal rehabilitation.
- Linke SE, Ussher M (January 2015). "Exercise-based treatments for substance use disorders: evidence, theory, and practicality". The American Journal of Drug and Alcohol Abuse. 41 (1): 7–15. doi:10.3109/00952990.2014.976708. PMC 4831948. PMID 25397661.
The limited research conducted suggests that exercise may be an effective adjunctive treatment for SUDs. In contrast to the scarce intervention trials to date, a relative abundance of literature on the theoretical and practical reasons supporting the investigation of this topic has been published. ... numerous theoretical and practical reasons support exercise-based treatments for SUDs, including psychological, behavioral, neurobiological, nearly universal safety profile, and overall positive health effects.
- Hyman SE, Malenka RC, Nestler EJ (July 2006). "Neural mechanisms of addiction: the role of reward-related learning and memory". Annual Review of Neuroscience. 29: 565–598. doi:10.1146/annurev.neuro.29.051605.113009. PMID 16776597.
- Steiner H, Van Waes V (January 2013). "Addiction-related gene regulation: risks of exposure to cognitive enhancers vs. other psychostimulants". Progress in Neurobiology. 100: 60–80. doi:10.1016/j.pneurobio.2012.10.001. PMC 3525776. PMID 23085425.
- Malenka RC, Nestler EJ, Hyman SE (2009). "Chapter 4: Signal Transduction in the Brain". In Sydor A, Brown RY (eds.). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York, USA: McGraw-Hill Medical. p. 94. ISBN 9780071481274.
- Kanehisa Laboratories (29 October 2014). "Alcoholism – Homo sapiens (human)". KEGG Pathway. Retrieved 31 October 2014.
- Kim Y, Teylan MA, Baron M, Sands A, Nairn AC, Greengard P (February 2009). "Methylphenidate-induced dendritic spine formation and DeltaFosB expression in nucleus accumbens". Proceedings of the National Academy of Sciences. 106 (8): 2915–2920. Bibcode:2009PNAS..106.2915K. doi:10.1073/pnas.0813179106. PMC 2650365. PMID 19202072.
- Nestler EJ (January 2014). "Epigenetic mechanisms of drug addiction". Neuropharmacology. 76 Pt B: 259–268. doi:10.1016/j.neuropharm.2013.04.004. PMC 3766384. PMID 23643695.
- Biliński P, Wojtyła A, Kapka-Skrzypczak L, Chwedorowicz R, Cyranka M, Studziński T (2012). "Epigenetic regulation in drug addiction". Annals of Agricultural and Environmental Medicine. 19 (3): 491–496. PMID 23020045.
- Kennedy PJ, Feng J, Robison AJ, Maze I, Badimon A, Mouzon E, Chaudhury D, Damez-Werno DM, Haggarty SJ, Han MH, Bassel-Duby R, Olson EN, Nestler EJ (April 2013). "Class I HDAC inhibition blocks cocaine-induced plasticity by targeted changes in histone methylation". Nature Neuroscience. 16 (4): 434–440. doi:10.1038/nn.3354. PMC 3609040. PMID 23475113.
- Whalley K (December 2014). "Psychiatric disorders: a feat of epigenetic engineering". Nature Reviews. Neuroscience. 15 (12): 768–769. doi:10.1038/nrn3869. PMID 25409693.
- Blum K, Werner T, Carnes S, Carnes P, Bowirrat A, Giordano J, Oscar-Berman M, Gold M (March 2012). "Sex, drugs, and rock 'n' roll: hypothesizing common mesolimbic activation as a function of reward gene polymorphisms". Journal of Psychoactive Drugs. 44 (1): 38–55. doi:10.1080/02791072.2012.662112. PMC 4040958. PMID 22641964.
It has been found that deltaFosB gene in the NAc is critical for reinforcing effects of sexual reward. Pitchers and colleagues (2010) reported that sexual experience was shown to cause DeltaFosB accumulation in several limbic brain regions including the NAc, medial pre-frontal cortex, VTA, caudate, and putamen, but not the medial preoptic nucleus. ... these findings support a critical role for DeltaFosB expression in the NAc in the reinforcing effects of sexual behavior and sexual experience-induced facilitation of sexual performance. ... both drug addiction and sexual addiction represent pathological forms of neuroplasticity along with the emergence of aberrant behaviors involving a cascade of neurochemical changes mainly in the brain's rewarding circuitry.
- Pitchers KK, Vialou V, Nestler EJ, Laviolette SR, Lehman MN, Coolen LM (February 2013). "Natural and drug rewards act on common neural plasticity mechanisms with ΔFosB as a key mediator". The Journal of Neuroscience. 33 (8): 3434–3442. doi:10.1523/JNEUROSCI.4881-12.2013. PMC 3865508. PMID 23426671.
- Beloate LN, Weems PW, Casey GR, Webb IC, Coolen LM (February 2016). "Nucleus accumbens NMDA receptor activation regulates amphetamine cross-sensitization and deltaFosB expression following sexual experience in male rats". Neuropharmacology. 101: 154–164. doi:10.1016/j.neuropharm.2015.09.023. PMID 26391065.
- Malenka RC, Nestler EJ, Hyman SE, Holtzman DM (2015). "Chapter 16: Reinforcement and Addictive Disorders". Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (3rd ed.). New York: McGraw-Hill Medical. ISBN 9780071827706.
Pharmacologic treatment for psychostimulant addiction is generally unsatisfactory. As previously discussed, cessation of cocaine use and the use of other psychostimulants in dependent individuals does not produce a physical withdrawal syndrome but may produce dysphoria, anhedonia, and an intense desire to reinitiate drug use.
- Chan B, Freeman M, Kondo K, Ayers C, Montgomery J, Paynter R, Kansagara D (December 2019). "Pharmacotherapy for methamphetamine/amphetamine use disorder-a systematic review and meta-analysis". Addiction (Abingdon, England). 114 (12): 2122–2136. doi:10.1111/add.14755. PMID 31328345.
- Stoops WW, Rush CR (May 2014). "Combination pharmacotherapies for stimulant use disorder: a review of clinical findings and recommendations for future research". Expert Review of Clinical Pharmacology. 7 (3): 363–374. doi:10.1586/17512433.2014.909283. PMC 4017926. PMID 24716825.
Despite concerted efforts to identify a pharmacotherapy for managing stimulant use disorders, no widely effective medications have been approved.
- Grandy DK, Miller GM, Li JX (February 2016). ""TAARgeting Addiction"-The Alamo Bears Witness to Another Revolution: An Overview of the Plenary Symposium of the 2015 Behavior, Biology and Chemistry Conference". Drug and Alcohol Dependence. 159: 9–16. doi:10.1016/j.drugalcdep.2015.11.014. PMC 4724540. PMID 26644139.
When considered together with the rapidly growing literature in the field a compelling case emerges in support of developing TAAR1-selective agonists as medications for preventing relapse to psychostimulant abuse.
- Jing L, Li JX (August 2015). "Trace amine-associated receptor 1: A promising target for the treatment of psychostimulant addiction". European Journal of Pharmacology. 761: 345–352. doi:10.1016/j.ejphar.2015.06.019. PMC 4532615. PMID 26092759.
Existing data provided robust preclinical evidence supporting the development of TAAR1 agonists as potential treatment for psychostimulant abuse and addiction.
- Malenka RC, Nestler EJ, Hyman SE (2009). "Chapter 5: Excitatory and Inhibitory Amino Acids". In Sydor A, Brown RY (eds.). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York, USA: McGraw-Hill Medical. pp. 124–125. ISBN 9780071481274.
- Carroll ME, Smethells JR (February 2016). "Sex Differences in Behavioral Dyscontrol: Role in Drug Addiction and Novel Treatments". Frontiers in Psychiatry. 6: 175. doi:10.3389/fpsyt.2015.00175. PMC 4745113. PMID 26903885.
Physical Exercise
There is accelerating evidence that physical exercise is a useful treatment for preventing and reducing drug addiction ... In some individuals, exercise has its own rewarding effects, and a behavioral economic interaction may occur, such that physical and social rewards of exercise can substitute for the rewarding effects of drug abuse. ... The value of this form of treatment for drug addiction in laboratory animals and humans is that exercise, if it can substitute for the rewarding effects of drugs, could be self-maintained over an extended period of time. Work to date in [laboratory animals and humans] regarding exercise as a treatment for drug addiction supports this hypothesis. ... Animal and human research on physical exercise as a treatment for stimulant addiction indicates that this is one of the most promising treatments on the horizon. - Perez-Mana C, Castells X, Torrens M, Capella D, Farre M (September 2013). "Efficacy of psychostimulant drugs for amphetamine abuse or dependence". Cochrane Database of Systematic Reviews. 9 (9): CD009695. doi:10.1002/14651858.CD009695.pub2. PMID 23996457.
- "Amphetamines: Drug Use and Abuse". Merck Manual Home Edition. Merck. February 2003. Archived from the original on 17 February 2007. Retrieved 28 February 2007.
- Shoptaw SJ, Kao U, Heinzerling K, Ling W (April 2009). Shoptaw SJ (ed.). "Treatment for amphetamine withdrawal". Cochrane Database of Systematic Reviews (2): CD003021. doi:10.1002/14651858.CD003021.pub2. PMC 7138250. PMID 19370579.
The prevalence of this withdrawal syndrome is extremely common (Cantwell 1998; Gossop 1982) with 87.6% of 647 individuals with amphetamine dependence reporting six or more signs of amphetamine withdrawal listed in the DSM when the drug is not available (Schuckit 1999) ... The severity of withdrawal symptoms is greater in amphetamine dependent individuals who are older and who have more extensive amphetamine use disorders (McGregor 2005). Withdrawal symptoms typically present within 24 hours of the last use of amphetamine, with a withdrawal syndrome involving two general phases that can last 3 weeks or more. The first phase of this syndrome is the initial "crash" that resolves within about a week (Gossop 1982;McGregor 2005) ...
- Spiller HA, Hays HL, Aleguas A (June 2013). "Overdose of drugs for attention-deficit hyperactivity disorder: clinical presentation, mechanisms of toxicity, and management". CNS Drugs. 27 (7): 531–543. doi:10.1007/s40263-013-0084-8. PMID 23757186.
Amphetamine, dextroamphetamine, and methylphenidate act as substrates for the cellular monoamine transporter, especially the dopamine transporter (DAT) and less so the norepinephrine (NET) and serotonin transporter. The mechanism of toxicity is primarily related to excessive extracellular dopamine, norepinephrine, and serotonin.
- Collaborators (2015). "Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013" (PDF). The Lancet. 385 (9963): 117–171. doi:10.1016/S0140-6736(14)61682-2. PMC 4340604. PMID 25530442. Retrieved 3 March 2015.
Amphetamine use disorders ... 3,788 (3,425–4,145)
- Greene SL, Kerr F, Braitberg G (October 2008). "Review article: amphetamines and related drugs of abuse". Emergency Medicine Australasia. 20 (5): 391–402. doi:10.1111/j.1742-6723.2008.01114.x. PMID 18973636.
- Albertson TE (2011). "Amphetamines". In Olson KR, Anderson IB, Benowitz NL, Blanc PD, Kearney TE, Kim-Katz SY, Wu AH (eds.). Poisoning & Drug Overdose (6th ed.). New York: McGraw-Hill Medical. pp. 77–79. ISBN 9780071668330.
- Advokat C (July 2007). "Update on amphetamine neurotoxicity and its relevance to the treatment of ADHD". Journal of Attention Disorders. 11 (1): 8–16. doi:10.1177/1087054706295605. PMID 17606768.
- Bowyer JF, Hanig JP (November 2014). "Amphetamine- and methamphetamine-induced hyperthermia: Implications of the effects produced in brain vasculature and peripheral organs to forebrain neurotoxicity". Temperature. 1 (3): 172–182. doi:10.4161/23328940.2014.982049. PMC 5008711. PMID 27626044.
Hyperthermia alone does not produce amphetamine-like neurotoxicity but AMPH and METH exposures that do not produce hyperthermia (≥40 °C) are minimally neurotoxic. Hyperthermia likely enhances AMPH and METH neurotoxicity directly through disruption of protein function, ion channels and enhanced ROS production. ... The hyperthermia and the hypertension produced by high doses amphetamines are a primary cause of transient breakdowns in the blood-brain barrier (BBB) resulting in concomitant regional neurodegeneration and neuroinflammation in laboratory animals. ... In animal models that evaluate the neurotoxicity of AMPH and METH, it is quite clear that hyperthermia is one of the essential components necessary for the production of histological signs of dopamine terminal damage and neurodegeneration in cortex, striatum, thalamus and hippocampus.
- "Amphetamine". United States National Library of Medicine – Toxicology Data Network. Hazardous Substances Data Bank. Archived from the original on 2 October 2017. Retrieved 2 October 2017.
Direct toxic damage to vessels seems unlikely because of the dilution that occurs before the drug reaches the cerebral circulation.
- Malenka RC, Nestler EJ, Hyman SE (2009). "Chapter 15: Reinforcement and addictive disorders". In Sydor A, Brown RY (eds.). Molecular Neuropharmacology: A Foundation for Clinical Neuroscience (2nd ed.). New York, USA: McGraw-Hill Medical. p. 370. ISBN 9780071481274.
Unlike cocaine and amphetamine, methamphetamine is directly toxic to midbrain dopamine neurons.
- Sulzer D, Zecca L (February 2000). "Intraneuronal dopamine-quinone synthesis: a review". Neurotoxicity Research. 1 (3): 181–195. doi:10.1007/BF03033289. PMID 12835101.
- Miyazaki I, Asanuma M (June 2008). "Dopaminergic neuron-specific oxidative stress caused by dopamine itself" (PDF). Acta Medica Okayama. 62 (3): 141–150. doi:10.18926/AMO/30942. PMID 18596830.
- Hofmann FG (1983). A Handbook on Drug and Alcohol Abuse: The Biomedical Aspects (2nd ed.). New York, USA: Oxford University Press. p. 329. ISBN 9780195030570.
- "Adderall XR Prescribing Information" (PDF). United States Food and Drug Administration. Shire US Inc. December 2013. pp. 8–10. Retrieved 30 December 2013.
- Miller GM (January 2011). "The emerging role of trace amine-associated receptor 1 in the functional regulation of monoamine transporters and dopaminergic activity". J. Neurochem. 116 (2): 164–176. doi:10.1111/j.1471-4159.2010.07109.x. PMC 3005101. PMID 21073468.
- Eiden LE, Weihe E (January 2011). "VMAT2: a dynamic regulator of brain monoaminergic neuronal function interacting with drugs of abuse". Ann. N. Y. Acad. Sci. 1216: 86–98. Bibcode:2011NYASA1216...86E. doi:10.1111/j.1749-6632.2010.05906.x. PMC 4183197. PMID 21272013.
VMAT2 is the CNS vesicular transporter for not only the biogenic amines DA, NE, EPI, 5-HT, and HIS, but likely also for the trace amines TYR, PEA, and thyronamine (THYR) ... [Trace aminergic] neurons in mammalian CNS would be identifiable as neurons expressing VMAT2 for storage, and the biosynthetic enzyme aromatic amino acid decarboxylase (AADC).
- Sulzer D, Cragg SJ, Rice ME (August 2016). "Striatal dopamine neurotransmission: regulation of release and uptake". Basal Ganglia. 6 (3): 123–148. doi:10.1016/j.baga.2016.02.001. PMC 4850498. PMID 27141430.
Despite the challenges in determining synaptic vesicle pH, the proton gradient across the vesicle membrane is of fundamental importance for its function. Exposure of isolated catecholamine vesicles to protonophores collapses the pH gradient and rapidly redistributes transmitter from inside to outside the vesicle. ... Amphetamine and its derivatives like methamphetamine are weak base compounds that are the only widely used class of drugs known to elicit transmitter release by a non-exocytic mechanism. As substrates for both DAT and VMAT, amphetamines can be taken up to the cytosol and then sequestered in vesicles, where they act to collapse the vesicular pH gradient.
- Ledonne A, Berretta N, Davoli A, Rizzo GR, Bernardi G, Mercuri NB (July 2011). "Electrophysiological effects of trace amines on mesencephalic dopaminergic neurons". Front. Syst. Neurosci. 5: 56. doi:10.3389/fnsys.2011.00056. PMC 3131148. PMID 21772817.
Three important new aspects of TAs action have recently emerged: (a) inhibition of firing due to increased release of dopamine; (b) reduction of D2 and GABAB receptor-mediated inhibitory responses (excitatory effects due to disinhibition); and (c) a direct TA1 receptor-mediated activation of GIRK channels which produce cell membrane hyperpolarization.
- "TAAR1". GenAtlas. University of Paris. 28 January 2012. Retrieved 29 May 2014.
• tonically activates inwardly rectifying K(+) channels, which reduces the basal firing frequency of dopamine (DA) neurons of the ventral tegmental area (VTA)
- Underhill SM, Wheeler DS, Li M, Watts SD, Ingram SL, Amara SG (July 2014). "Amphetamine modulates excitatory neurotransmission through endocytosis of the glutamate transporter EAAT3 in dopamine neurons". Neuron. 83 (2): 404–416. doi:10.1016/j.neuron.2014.05.043. PMC 4159050. PMID 25033183.
AMPH also increases intracellular calcium (Gnegy et al., 2004) that is associated with calmodulin/CamKII activation (Wei et al., 2007) and modulation and trafficking of the DAT (Fog et al., 2006; Sakrikar et al., 2012). ... For example, AMPH increases extracellular glutamate in various brain regions including the striatum, VTA and NAc (Del Arco et al., 1999; Kim et al., 1981; Mora and Porras, 1993; Xue et al., 1996), but it has not been established whether this change can be explained by increased synaptic release or by reduced clearance of glutamate. ... DHK-sensitive, EAAT2 uptake was not altered by AMPH (Figure 1A). The remaining glutamate transport in these midbrain cultures is likely mediated by EAAT3 and this component was significantly decreased by AMPH
- Vaughan RA, Foster JD (September 2013). "Mechanisms of dopamine transporter regulation in normal and disease states". Trends Pharmacol. Sci. 34 (9): 489–496. doi:10.1016/j.tips.2013.07.005. PMC 3831354. PMID 23968642.
AMPH and METH also stimulate DA efflux, which is thought to be a crucial element in their addictive properties [80], although the mechanisms do not appear to be identical for each drug [81]. These processes are PKCβ– and CaMK–dependent [72, 82], and PKCβ knock-out mice display decreased AMPH-induced efflux that correlates with reduced AMPH-induced locomotion [72].
- "Identification". Lisdexamfetamine. DrugBank. University of Alberta. 16 September 2013. Retrieved 13 June 2014.
- Jasinski DR, Krishnan S (June 2009). "Abuse liability and safety of oral lisdexamfetamine dimesylate in individuals with a history of stimulant abuse". J. Psychopharmacol. (Oxford). 23 (4): 419–427. doi:10.1177/0269881109103113. PMID 19329547.
- "Adderall XR Prescribing Information" (PDF). United States Food and Drug Administration. pp. 1–18. Retrieved 7 October 2013.
- "Pharmacology". Dextroamphetamine. DrugBank. University of Alberta. 8 February 2013. Retrieved 5 November 2013.
- "Adderall XR Prescribing Information" (PDF). United States Food and Drug Administration. Shire US Inc. December 2013. pp. 12–13. Retrieved 30 December 2013.
- "Pharmacology". Amphetamine. DrugBank. University of Alberta. 8 February 2013. Retrieved 5 November 2013.
- "Metabolism/Pharmacokinetics". Amphetamine. United States National Library of Medicine – Toxicology Data Network. Hazardous Substances Data Bank. Archived from the original on 2 October 2017. Retrieved 2 October 2017.
Duration of effect varies depending on agent and urine pH. Excretion is enhanced in more acidic urine. Half-life is 7 to 34 hours and is, in part, dependent on urine pH (half-life is longer with alkaline urine). ... Amphetamines are distributed into most body tissues with high concentrations occurring in the brain and CSF. Amphetamine appears in the urine within about 3 hours following oral administration. ... Three days after a dose of (+ or -)-amphetamine, human subjects had excreted 91% of the (14)C in the urine
- Santagati NA, Ferrara G, Marrazzo A, Ronsisvalle G (September 2002). "Simultaneous determination of amphetamine and one of its metabolites by HPLC with electrochemical detection". Journal of Pharmaceutical and Biomedical Analysis. 30 (2): 247–255. doi:10.1016/S0731-7085(02)00330-8. PMID 12191709.
- "Compound Summary". p-Hydroxyamphetamine. PubChem Compound Database. United States National Library of Medicine – National Center for Biotechnology Information. Retrieved 15 October 2013.
- "Compound Summary". p-Hydroxynorephedrine. PubChem Compound Database. United States National Library of Medicine – National Center for Biotechnology Information. Retrieved 15 October 2013.
- "Compound Summary". Phenylpropanolamine. PubChem Compound Database. United States National Library of Medicine – National Center for Biotechnology Information. Retrieved 15 October 2013.
- "Pharmacology and Biochemistry". Amphetamine. Pubchem Compound Database. United States National Library of Medicine – National Center for Biotechnology Information. Retrieved 12 October 2013.
- Glennon RA (2013). "Phenylisopropylamine stimulants: amphetamine-related agents". In Lemke TL, Williams DA, Roche VF, Zito W (eds.). Foye's principles of medicinal chemistry (7th ed.). Philadelphia, USA: Wolters Kluwer Health/Lippincott Williams & Wilkins. pp. 646–648. ISBN 9781609133450.
The simplest unsubstituted phenylisopropylamine, 1-phenyl-2-aminopropane, or amphetamine, serves as a common structural template for hallucinogens and psychostimulants. Amphetamine produces central stimulant, anorectic, and sympathomimetic actions, and it is the prototype member of this class (39). ... The phase 1 metabolism of amphetamine analogs is catalyzed by two systems: cytochrome P450 and flavin monooxygenase. ... Amphetamine can also undergo aromatic hydroxylation to p-hydroxyamphetamine. ... Subsequent oxidation at the benzylic position by DA β-hydroxylase affords p-hydroxynorephedrine. Alternatively, direct oxidation of amphetamine by DA β-hydroxylase can afford norephedrine.
- Taylor KB (January 1974). "Dopamine-beta-hydroxylase. Stereochemical course of the reaction" (PDF). Journal of Biological Chemistry. 249 (2): 454–458. PMID 4809526. Retrieved 6 November 2014.
Dopamine-β-hydroxylase catalyzed the removal of the pro-R hydrogen atom and the production of 1-norephedrine, (2S,1R)-2-amino-1-hydroxyl-1-phenylpropane, from d-amphetamine.
- Krueger SK, Williams DE (June 2005). "Mammalian flavin-containing monooxygenases: structure/function, genetic polymorphisms and role in drug metabolism". Pharmacology & Therapeutics. 106 (3): 357–387. doi:10.1016/j.pharmthera.2005.01.001. PMC 1828602. PMID 15922018.
Table 5: N-containing drugs and xenobiotics oxygenated by FMO - Cashman JR, Xiong YN, Xu L, Janowsky A (March 1999). "N-oxygenation of amphetamine and methamphetamine by the human flavin-containing monooxygenase (form 3): role in bioactivation and detoxication". Journal of Pharmacology and Experimental Therapeutics. 288 (3): 1251–1260. PMID 10027866.
- Sjoerdsma A, von Studnitz W (April 1963). "Dopamine-beta-oxidase activity in man, using hydroxyamphetamine as substrate". British Journal of Pharmacology and Chemotherapy. 20: 278–284. doi:10.1111/j.1476-5381.1963.tb01467.x. PMC 1703637. PMID 13977820.
Hydroxyamphetamine was administered orally to five human subjects ... Since conversion of hydroxyamphetamine to hydroxynorephedrine occurs in vitro by the action of dopamine-β-oxidase, a simple method is suggested for measuring the activity of this enzyme and the effect of its inhibitors in man. ... The lack of effect of administration of neomycin to one patient indicates that the hydroxylation occurs in body tissues. ... a major portion of the β-hydroxylation of hydroxyamphetamine occurs in non-adrenal tissue. Unfortunately, at the present time one cannot be completely certain that the hydroxylation of hydroxyamphetamine in vivo is accomplished by the same enzyme which converts dopamine to noradrenaline.
- Badenhorst CP, van der Sluis R, Erasmus E, van Dijk AA (September 2013). "Glycine conjugation: importance in metabolism, the role of glycine N-acyltransferase, and factors that influence interindividual variation". Expert Opinion on Drug Metabolism & Toxicology. 9 (9): 1139–1153. doi:10.1517/17425255.2013.796929. PMID 23650932.
Figure 1. Glycine conjugation of benzoic acid. The glycine conjugation pathway consists of two steps. First benzoate is ligated to CoASH to form the high-energy benzoyl-CoA thioester. This reaction is catalyzed by the HXM-A and HXM-B medium-chain acid:CoA ligases and requires energy in the form of ATP. ... The benzoyl-CoA is then conjugated to glycine by GLYAT to form hippuric acid, releasing CoASH. In addition to the factors listed in the boxes, the levels of ATP, CoASH, and glycine may influence the overall rate of the glycine conjugation pathway.
- Horwitz D, Alexander RW, Lovenberg W, Keiser HR (May 1973). "Human serum dopamine-β-hydroxylase. Relationship to hypertension and sympathetic activity". Circulation Research. 32 (5): 594–599. doi:10.1161/01.RES.32.5.594. PMID 4713201.
The biologic significance of the different levels of serum DβH activity was studied in two ways. First, in vivo ability to β-hydroxylate the synthetic substrate hydroxyamphetamine was compared in two subjects with low serum DβH activity and two subjects with average activity. ... In one study, hydroxyamphetamine (Paredrine), a synthetic substrate for DβH, was administered to subjects with either low or average levels of serum DβH activity. The percent of the drug hydroxylated to hydroxynorephedrine was comparable in all subjects (6.5-9.62) (Table 3).
- Freeman JJ, Sulser F (December 1974). "Formation of p-hydroxynorephedrine in brain following intraventricular administration of p-hydroxyamphetamine". Neuropharmacology. 13 (12): 1187–1190. doi:10.1016/0028-3908(74)90069-0. PMID 4457764.
In species where aromatic hydroxylation of amphetamine is the major metabolic pathway, p-hydroxyamphetamine (POH) and p-hydroxynorephedrine (PHN) may contribute to the pharmacological profile of the parent drug. ... The location of the p-hydroxylation and β-hydroxylation reactions is important in species where aromatic hydroxylation of amphetamine is the predominant pathway of metabolism. Following systemic administration of amphetamine to rats, POH has been found in urine and in plasma.
The observed lack of a significant accumulation of PHN in brain following the intraventricular administration of (+)-amphetamine and the formation of appreciable amounts of PHN from (+)-POH in brain tissue in vivo supports the view that the aromatic hydroxylation of amphetamine following its systemic administration occurs predominantly in the periphery, and that POH is then transported through the blood-brain barrier, taken up by noradrenergic neurones in brain where (+)-POH is converted in the storage vesicles by dopamine β-hydroxylase to PHN. - Matsuda LA, Hanson GR, Gibb JW (December 1989). "Neurochemical effects of amphetamine metabolites on central dopaminergic and serotonergic systems". Journal of Pharmacology and Experimental Therapeutics. 251 (3): 901–908. PMID 2600821.
The metabolism of p-OHA to p-OHNor is well documented and dopamine-β hydroxylase present in noradrenergic neurons could easily convert p-OHA to p-OHNor after intraventricular administration.
- "Lidsexamfetamine". ChemSpider. Royal Society of Chemistry. 2015. Retrieved 22 April 2019.
- Stahl, P. Heinrich; Wermuth, Camille G., eds. (2011). Pharmaceutical Salts: Properties, Selection, and Use (2nd ed.). John Wiley & Sons. ISBN 978-3-90639-051-2.
- "Molecular Weight Calculator". Lenntech. Retrieved 19 August 2015.
- "Dextroamphetamine Sulfate USP". Mallinckrodt Pharmaceuticals. March 2014. Retrieved 19 August 2015.
- "D-amphetamine sulfate". Tocris. 2015. Retrieved 19 August 2015.
- "Amphetamine Sulfate USP". Mallinckrodt Pharmaceuticals. March 2014. Retrieved 19 August 2015.
- "Dextroamphetamine Saccharate". Mallinckrodt Pharmaceuticals. March 2014. Retrieved 19 August 2015.
- "Amphetamine Aspartate". Mallinckrodt Pharmaceuticals. March 2014. Retrieved 19 August 2015.
- Mattingly, G (May 2010). "Lisdexamfetamine dimesylate: a prodrug stimulant for the treatment of ADHD in children and adults". CNS Spectrums. 15 (5): 315–25. doi:10.1017/S1092852900027541. PMID 20448522.
- "FDA Adult Approval of Vyvanse – FDA Label and Approval History" (PDF).
- Health Canada Notice of Compliance – Vyvanse. 19 February 2009, retrieved on 9 March 2009.
- "Press Announcements – FDA expands uses of Vyvanse to treat binge-eating disorder". FDA. 30 January 2015.
- "DEA Office of Diversion Control" (PDF). DEA. Retrieved 1 July 2014.
- "Lisdexamfetamine international brands". Drugs.com. Retrieved 10 July 2017.
- Dale E, Bang-Andersen B, Sánchez C (May 2015). "Emerging mechanisms and treatments for depression beyond SSRIs and SNRIs". Biochem. Pharmacol. 95 (2): 81–97. doi:10.1016/j.bcp.2015.03.011. PMID 25813654.
- Stotz, G.; Woggon, B.; Angst, J. (1999). "Psychostimulants in the therapy of treatment-resistant depression Review of the literature and findings from a retrospective study in 65 depressed patients". Dialogues Clin. Neurosci. 1 (3): 165–74. PMC 3181580. PMID 22034135.
- Hirschler, Ben (7 February 2014). "UPDATE 2-Shire scraps Vyvanse for depression after failed trials". Reuters. Retrieved 13 February 2014.
External links
- "Lisdexamfetamine". Drug Information Portal. U.S. National Library of Medicine.