Pyridine
Pyridine is a basic heterocyclic organic compound with the chemical formula C5H5N. It is structurally related to benzene, with one methine group (=CH−) replaced by a nitrogen atom. It is a highly flammable, weakly alkaline, water-miscible liquid with a distinctive, unpleasant fish-like smell. Pyridine is colorless, but older or impure samples can appear yellow. The pyridine ring occurs in many important compounds, including agrochemicals, pharmaceuticals, and vitamins. Historically, pyridine was produced from coal tar. Today it is synthesized on the scale of about 20,000 tonnes per year worldwide.[2]
| |||
| |||
 | |||
| Names | |||
|---|---|---|---|
| Preferred IUPAC name
Pyridine[1] | |||
| Systematic IUPAC name
Azabenzene | |||
| Other names
Azine Azinine 1-Azacyclohexa-1,3,5-diene | |||
| Identifiers | |||
3D model (JSmol) |
|||
| ChEBI | |||
| ChEMBL | |||
| ChemSpider | |||
| ECHA InfoCard | 100.003.464 | ||
| EC Number |
| ||
| KEGG | |||
PubChem CID |
|||
| UNII | |||
CompTox Dashboard (EPA) |
|||
| |||
| |||
| Properties | |||
| C5H5N | |||
| Molar mass | 79.102 g·mol−1 | ||
| Appearance | Colorless liquid[2] | ||
| Odor | Nauseating, fish-like[3] | ||
| Density | 0.9819 g/mL[4] | ||
| Melting point | −41.6 °C (−42.9 °F; 231.6 K) | ||
| Boiling point | 115.2 °C (239.4 °F; 388.3 K) | ||
| Miscible | |||
| log P | 0.73 [5] | ||
| Vapor pressure | 16 mmHg (20 °C)[3] | ||
| Conjugate acid | Pyridinium | ||
Refractive index (nD) |
1.5093 | ||
| Viscosity | 0.88 cP 25℃ | ||
| 2.2 D[6] | |||
| Hazards[7] | |||
| Safety data sheet | See: data page | ||
EU classification (DSD) (outdated) |
Flammable (F) Harmful (Xn) | ||
| R-phrases (outdated) | R20 R21 R22 R34 R36 R38 | ||
| NFPA 704 (fire diamond) | |||
| Flash point | 21 °C (70 °F; 294 K) | ||
| Explosive limits | 1.8–12.4%[3] | ||
Threshold limit value (TLV) |
5 ppm (TWA) | ||
| Lethal dose or concentration (LD, LC): | |||
LD50 (median dose) |
891 mg/kg (rat, oral) 1500 mg/kg (mouse, oral) 1580 mg/kg (rat, oral)[8] | ||
LC50 (median concentration) |
9000 ppm (rat, 1 hr)[8] | ||
| NIOSH (US health exposure limits): | |||
PEL (Permissible) |
TWA 5 ppm (15 mg/m3)[3] | ||
REL (Recommended) |
TWA 5 ppm (15 mg/m3)[3] | ||
IDLH (Immediate danger) |
1000 ppm[3] | ||
| Related compounds | |||
Related amines |
Picoline Quinoline | ||
Related compounds |
Aniline Pyrimidine Piperidine | ||
| Supplementary data page | |||
| Refractive index (n), Dielectric constant (εr), etc. | |||
Thermodynamic data |
Phase behaviour solid–liquid–gas | ||
| UV, IR, NMR, MS | |||
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). | |||
| Infobox references | |||

Properties
Physical properties
The molecular electric dipole moment is 2.2 debyes.[6] Pyridine is diamagnetic and has a diamagnetic susceptibility of −48.7 × 10−6 cm3·mol−1.[9] The standard enthalpy of formation is 100.2 kJ·mol−1 in the liquid phase[10] and 140.4 kJ·mol−1 in the gas phase. At 25 °C pyridine has a viscosity[11] of 0.88 mPa/s and thermal conductivity of 0.166 W·m−1·K−1.[12][13] The enthalpy of vaporization is 35.09 kJ·mol−1 at the boiling point and normal pressure.[14] The enthalpy of fusion is 8.28 kJ·mol−1 at the melting point.[15]
The critical parameters of pyridine are pressure 6.70 MPa, temperature 620 K and volume 229 cm3·mol−1.[16] In the temperature range 340–426 °C its vapor pressure p can be described with the Antoine equation

where T is temperature, A = 4.16272, B = 1371.358 K and C = −58.496 K.[17]
Structure
Akin to benzene, pyridine ring forms a C5N hexagon. Electron localization in pyridine is reflected in the shorter C–N ring bond (137 pm for the C–N bond in pyridine vs. 139 pm for C–C bond in benzene),[18] whereas the carbon–carbon bonds in the pyridine ring have the same 139 pm length as in benzene. These bond lengths lie between the values for the single and double bonds and are typical of aromatic compounds.
Crystallography
Pyridine crystallizes in an orthorhombic crystal system with space group Pna21 and lattice parameters a = 1752 pm, b = 897 pm, c = 1135 pm, and 16 formula units per unit cell (measured at 153 K). For comparison, crystalline benzene is also orthorhombic, with space group Pbca, a = 729.2 pm, b = 947.1 pm, c = 674.2 pm (at 78 K), but the number of molecules per cell is only 4.[19] This difference is partly related to the lower symmetry of the individual pyridine molecule (C2v vs D6h for benzene). A trihydrate (pyridine·3H2O) is known; it also crystallizes in an orthorhombic system in the space group Pbca, lattice parameters a = 1244 pm, b = 1783 pm, c = 679 pm and eight formula units per unit cell (measured at 223 K).[20]
Spectroscopy
The optical absorption spectrum of pyridine in hexane contains three bands at the wavelengths of 195 nm (π → π* transition, molar absorptivity ε = 7500 L·mol−1·cm−1), 251 nm (π → π* transition, ε = 2000 L·mol−1·cm−1) and 270 nm (n → π* transition, ε = 450 L·mol−1·cm−1).[21] The 1H nuclear magnetic resonance (NMR) spectrum of pyridine contains three signals with the integral intensity ratio of 2:1:2 that correspond to the three chemically different protons in the molecule. These signals originate from the α-protons (positions 2 and 6, chemical shift 8.5 ppm), γ-proton (position 4, 7.5 ppm) and β-protons (positions 3 and 5, 7.1 ppm). The carbon analog of pyridine, benzene, has only one proton signal at 7.27 ppm. The larger chemical shifts of the α- and γ-protons in comparison to benzene result from the lower electron density in the α- and γ-positions, which can be derived from the resonance structures. The situation is rather similar for the 13C NMR spectra of pyridine and benzene: pyridine shows a triplet at δ(α-C) = 150 ppm, δ(β-C) = 124 ppm and δ(γ-C) = 136 ppm, whereas benzene has a single line at 129 ppm. All shifts are quoted for the solvent-free substances.[22] Pyridine is conventionally detected by the gas chromatography and mass spectrometry methods.[23]
Chemical properties
Because of the electronegative nitrogen in the pyridine ring , the molecule is relatively electron deficient. It, therefore, enters less readily into electrophilic aromatic substitution reactions than benzene derivatives. Correspondingly pyridine is more prone to nucleophilic substitution, as evidenced by the ease of metalation by strong organometallic bases.[24][25] The reactivity of pyridine can be distinguished for three chemical groups. With electrophiles, electrophilic substitution takes place where pyridine expresses aromatic properties. With nucleophiles, pyridine reacts at positions 2 and 4 and thus behaves similar to imines and carbonyls. The reaction with many Lewis acids results in the addition to the nitrogen atom of pyridine, which is similar to the reactivity of tertiary amines. The ability of pyridine and its derivatives to oxidize, forming amine oxides (N-oxides), is also a feature of tertiary amines.[26]
The nitrogen center of pyridine features a basic lone pair of electrons. This lone pair does not overlap with the aromatic π-system ring, consequently pyridine is basic, having chemical properties similar to those of tertiary amines. Protonation gives pyridinium, C5H5NH+.The pKa of the conjugate acid (the pyridinium cation) is 5.25. The structures of pyridine and pyridinium are almost identical.[27] The pyridinium cation is isoelectronic with benzene. Pyridinium p-toluenesulfonate (PPTS) is an illustrative pyridinium salt; it is produced by treating pyridine with p-toluenesulfonic acid. In addition to protonation, pyridine undergoes N-centered alkylation, acylation, and N-oxidation.
Bonding
Pyridine has a conjugated system of six π electrons that are delocalized over the ring. The molecule is planar and, thus, follows the Hückel criteria for aromatic systems. In contrast to benzene, the electron density is not evenly distributed over the ring, reflecting the negative inductive effect of the nitrogen atom. For this reason, pyridine has a dipole moment and a weaker resonant stabilization than benzene (resonance energy 117 kJ·mol−1 in pyridine vs. 150 kJ·mol−1 in benzene).[28]
The ring atoms in the pyridine molecule are sp2-hybridized. The nitrogen is involved in the π-bonding aromatic system using its unhybridized p orbital. The lone pair is in an sp2 orbital, projecting outward from the ring in the same plane as the σ bonds. As a result, the lone pair does not contribute to the aromatic system but importantly influences the chemical properties of pyridine, as it easily supports bond formation via an electrophilic attack. However, because of the separation of the lone pair from the aromatic ring system, the nitrogen atom cannot exhibit a positive mesomeric effect.
Many analogues of pyridine are known where N is replaced by other heteroatoms (see figure below). Substitution of one C–H in pyridine with a second N gives rise to the diazine heterocycles (C4H4N2), with the names pyridazine, pyrimidine, and pyrazine.
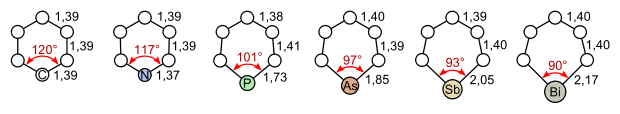 Bond lengths and angles of benzene, pyridine, phosphorine, arsabenzene, stibabenzene, and bismabenzene |
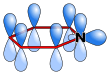 Atomic orbitals in pyridine |
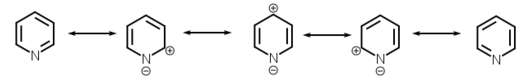 Resonance structures of pyridine |
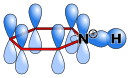 Atomic orbitals in protonated pyridine |
History
.jpg)
Impure pyridine was undoubtedly prepared by early alchemists by heating animal bones and other organic matter,[29] but the earliest documented reference is attributed to the Scottish scientist Thomas Anderson.[30][31] In 1849, Anderson examined the contents of the oil obtained through high-temperature heating of animal bones.[31] Among other substances, he separated from the oil a colorless liquid with unpleasant odor, from which he isolated pure pyridine two years later. He described it as highly soluble in water, readily soluble in concentrated acids and salts upon heating, and only slightly soluble in oils.
Owing to its flammability, Anderson named the new substance pyridine, after Greek: πῦρ (pyr) meaning fire. The suffix idine was added in compliance with the chemical nomenclature, as in toluidine, to indicate a cyclic compound containing a nitrogen atom.[32][33]
The chemical structure of pyridine was determined decades after its discovery. Wilhelm Körner (1869)[34] and James Dewar (1871)[35][36] suggested that, in analogy between quinoline and naphthalene, the structure of pyridine is derived from benzene by substituting one C–H unit with a nitrogen atom.[37][38] The suggestion by Körner and Dewar was later confirmed in an experiment where pyridine was reduced to piperidine with sodium in ethanol.[39] In 1876, William Ramsay combined acetylene and hydrogen cyanide into pyridine in a red-hot iron-tube furnace.[40] This was the first synthesis of a heteroaromatic compound.[23][41]
The first major synthesis of pyridine derivatives was described in 1881 by Arthur Rudolf Hantzsch.[42] The Hantzsch pyridine synthesis typically uses a 2:1:1 mixture of a β-keto acid (often acetoacetate), an aldehyde (often formaldehyde), and ammonia or its salt as the nitrogen donor. First, a double hydrogenated pyridine is obtained, which is then oxidized to the corresponding pyridine derivative. Emil Knoevenagel showed that asymmetrically-substituted pyridine derivatives can be produced with this process.[43]
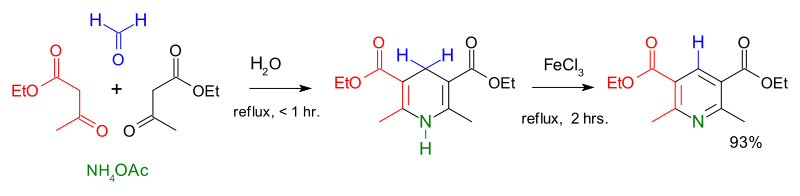
The contemporary methods of pyridine production had a low yield, and the increasing demand for the new compound urged to search for more efficient routes. A breakthrough came in 1924 when the Russian chemist Aleksei Chichibabin invented a pyridine synthesis reaction, which was based on inexpensive reagents.[44] This method is still used for the industrial production of pyridine.[2]
Occurrence
Pyridine is not abundant in nature, except for the leaves and roots of belladonna (Atropa belladonna)[45] and in marshmallow (Althaea officinalis).[46] Pyridine derivatives, however, are often part of biomolecules such as alkaloids.
In daily life, trace amounts of pyridine are components of the volatile organic compounds that are produced in roasting and canning processes, e.g. in fried chicken,[47] sukiyaki,[48] roasted coffee,[49] potato chips,[50] and fried bacon.[51] Traces of pyridine can be found in Beaufort cheese,[52] vaginal secretions,[53] black tea,[54] saliva of those suffering from gingivitis,[55] and sunflower honey.[56]
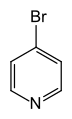 4-bromopyridine
4-bromopyridine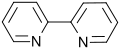 2,2′-bipyridine
2,2′-bipyridine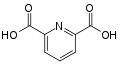 pyridine-2,6-dicarboxylic acid (dipicolinic acid)
pyridine-2,6-dicarboxylic acid (dipicolinic acid)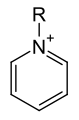 General form of the pyridinium cation
General form of the pyridinium cation
Production
Historically, pyridine was extracted from coal tar or obtained as a byproduct of coal gasification. The process was labor-consuming and inefficient: coal tar contains only about 0.1% pyridine,[57] and therefore a multi-stage purification was required, which further reduced the output. Nowadays, most pyridine is produced synthetically using various name reactions, and the major ones are discussed below.[2]
In 1989, 26,000 tonnes of pyridine was produced worldwide.[2] Among the largest 25 production sites for pyridine, eleven are located in Europe (as of 1999).[23] The major producers of pyridine include Evonik Industries, Rütgers Chemicals, Jubilant Life Sciences, Imperial Chemical Industries, and Koei Chemical.[2] Pyridine production significantly increased in the early 2000s, with an annual production capacity of 30,000 tonnes in mainland China alone.[58] The US–Chinese joint venture Vertellus is currently the world leader in pyridine production.[59]
Chichibabin synthesis
The Chichibabin pyridine synthesis was reported in 1924 and is still in use in industry.[44] In its general form, the reaction can be described as a condensation reaction of aldehydes, ketones, α,β-unsaturated carbonyl compounds, or any combination of the above, in ammonia or ammonia derivatives.[60] In particular, unsubstituted pyridine is produced from formaldehyde and acetaldehyde, which are inexpensive and widely available. First, acrolein is formed in a Knoevenagel condensation from the acetaldehyde and formaldehyde. The acrolein is then condensed with acetaldehyde and ammonia to give dihydropyridine, which is oxidized with a solid-state catalyst to pyridine. This process is carried out in a gas phase at 400–450 °C. The product consists of a mixture of pyridine, simple methylated pyridines (picolines and lutidines); its composition depends on the catalyst used and can be adapted to the needs of the manufacturer. The catalyst is usually a transition metal salt such as cadmium(II) fluoride or manganese(II) fluoride, but cobalt and thallium compounds can also be used. The recovered pyridine is separated from byproducts in a multistage process.[2]

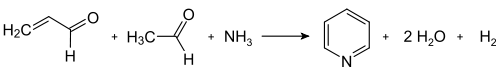
Practical application of the traditional Chichibabin pyridine synthesis are limited by its consistently low yield, typically about 20%. This low yield, together with the high prevalence of byproducts, render unmodified forms of Chichibabin's method unpopular.[60]
Dealkylation of alkylpyridines
Pyridine can be prepared by dealkylation of alkylated pyridines, which are obtained as byproducts in the syntheses of other pyridines. The oxidative dealkylation is carried out either using air over vanadium(V) oxide catalyst,[61] by vapor-dealkylation on nickel-based catalyst,[62][63] or hydrodealkylation with a silver- or platinum-based catalyst.[64] Yields of pyridine up to be 93% can be achieved with the nickel-based catalyst.[2]
Bönnemann cyclization

The trimerization of a part of a nitrile molecule and two parts of acetylene into pyridine is called Bönnemann cyclization. This modification of the Reppe synthesis can be activated either by heat or by light. While the thermal activation requires high pressures and temperatures, the photoinduced cycloaddition proceeds at ambient conditions with CoCp2(cod) (Cp = cyclopentadienyl, cod = 1,5-cyclooctadiene) as a catalyst, and can be performed even in water.[65] A series of pyridine derivatives can be produced in this way. When using acetonitrile as the nitrile, 2-methylpyridine is obtained, which can be dealkylated to pyridine.
Other methods
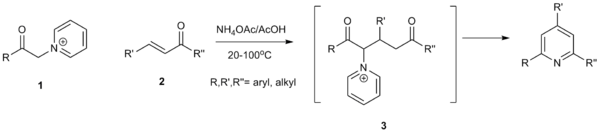
The Kröhnke pyridine synthesis provides a fairly general method for generating substituted pyridines using pyridine itself as a reagent which does not become incorporated into the final product. The reaction of pyridine with α-bromoesters give the related pyridinium salt, wherein the methylene group is highly acidic. This species undergoes a Michael-like addition to α,β-unsaturated carbonyls in the presence of ammonium acetate to undergo ring closure and formation of the targeted substituted pyridine as well as pyridinium bromide.[66]
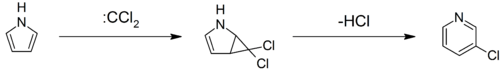
The Ciamician–Dennstedt rearrangement entails the ring-expansion of pyrrole with dichlorocarbene to 3-chloropyridine.[67][68][69]
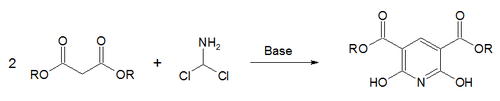
In the Gattermann–Skita synthesis,[70] a malonate ester salt reacts with dichloromethylamine.[71]
Another method is the Boger pyridine synthesis.
Pyridine can also be produced by the decarboxylation of nicotinic acid with copper chromite.[72]
Biosynthesis
Several pyridine derivatives play important roles in biological systems. While its biosynthesis is not fully understood, nicotinic acid (vitamin B3) occurs in some bacteria, fungi, and mammals. Mammals synthesize nicotinic acid through oxidation of the amino acid tryptophan, where an intermediate product, aniline, creates a pyridine derivative, kynurenine. On the contrary, the bacteria Mycobacterium tuberculosis and Escherichia coli produce nicotinic acid by condensation of glyceraldehyde 3-phosphate and aspartic acid.[73]
Reactions
Despite the structural and bonding commonalities of benzene and pyridine, their reactivity differ significantly. Instead, in terms of its reactivity, pyridine more closely resembles nitrobenzene.[74]
Electrophilic substitutions
Owing to the decreased electron density in the aromatic system, electrophilic substitutions are suppressed in pyridine and its derivatives. Friedel–Crafts alkylation or acylation, usually fail for pyridine because they lead only to the addition at the nitrogen atom. Substitutions usually occur at the 3-position, which is the most electron-rich carbon atom in the ring and is, therefore, more susceptible to an electrophilic addition.
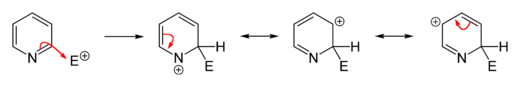
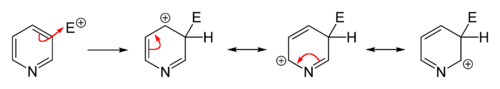
Direct nitration of pyridine is sluggish.[75][76] Pyridine derivatives wherein the nitrogen atom is screened sterically and/or electronically can be obtained by nitration with nitronium tetrafluoroborate (NO2BF4). In this way, 3-nitropyridine can be obtained via the synthesis of 2,6-dibromopyridine followed by debromination.[77][78]
Sulfonation of pyridine is even more difficult than nitration. However, pyridine-3-sulfonic acid can be obtained. Reaction with the SO3 group also facilitates addition of sulfur to the nitrogen atom, especially in the presence of a mercury(II) sulfate catalyst.[24][79]
In contrast to the sluggish nitrations and sulfonations, the bromination and chlorination of pyridine proceed well.[2]

Pyridine-N-oxide
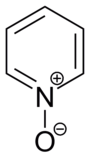
Oxidation of pyridine occurs at nitrogen to give pyridine-N-oxide. The oxidation can be achieved with peracids:[80]
- C5H5N + RCO3H → C5H5NO + RCO2H
Some electrophilic substitutions on the pyridine are usefully effected using pyridine-N-oxide followed by deoxygenation. Addition of oxygen suppresses further reactions at nitrogen atom and promotes substitution at the 2- and 4-carbons. The oxygen atom can then be removed, e.g. using zinc dust.[81]
Nucleophilic substitutions
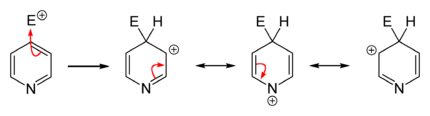
In contrast to benzene ring, pyridine efficiently supports several nucleophilic substitutions. The reason for this is relatively lower electron density of the carbon atoms of the ring. These reactions include substitutions with elimination of a hydride ion and elimination-additions with formation of an intermediate aryne configuration, and usually proceed at the 2- or 4-position.[24][25]
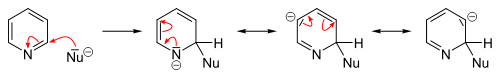
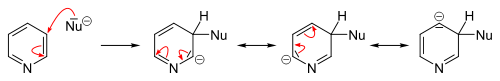
Many nucleophilic substitutions occur more easily not with bare pyridine but with pyridine modified with bromine, chlorine, fluorine, or sulfonic acid fragments that then become a leaving group. So fluorine is the best leaving group for the substitution with organolithium compounds. The nucleophilic attack compounds may be alkoxides, thiolates, amines, and ammonia (at elevated pressures).[82]
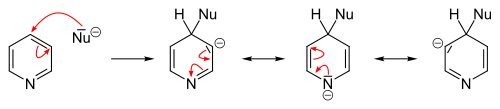
In general, the hydride ion is a poor leaving group and occurs only in a few heterocyclic reactions. They include the Chichibabin reaction, which yields pyridine derivatives aminated at the 2-position. Here, sodium amide is used as the nucleophile yielding 2-aminopyridine. The hydride ion released in this reaction combines with a proton of an available amino group, forming a hydrogen molecule.[25][83]
Analogous to benzene, nucleophilic substitutions to pyridine can result in the formation of pyridyne intermediates as heteroaryne. For this purpose, pyridine derivatives can be eliminated with good leaving groups using strong bases such as sodium and potassium tert-butoxide. The subsequent addition of a nucleophile to the triple bond has low selectivity, and the result is a mixture of the two possible adducts.[24]
Radical reactions
Pyridine supports a series of radical reactions, which is used in its dimerization to bipyridines. Radical dimerization of pyridine with elemental sodium or Raney nickel selectively yields 4,4′-bipyridine,[84] or 2,2′-bipyridine,[85] which are important precursor reagents in the chemical industry. One of the name reactions involving free radicals is the Minisci reaction. It can produce 2-tert-butylpyridine upon reacting pyridine with pivalic acid, silver nitrate and ammonium in sulfuric acid with a yield of 97%.[24]
Reactions on the nitrogen atom
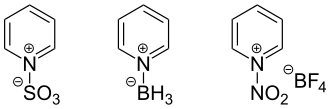
Lewis acids easily add to the nitrogen atom of pyridine, forming pyridinium salts. The reaction with alkyl halides leads to alkylation of the nitrogen atom. This creates a positive charge in the ring that increases the reactivity of pyridine to both oxidation and reduction. The Zincke reaction is used for the selective introduction of radicals in pyridinium compounds (it has no relation to the chemical element zinc).
Hydrogenation and reduction
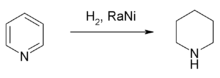
Piperidine is produced by hydrogenation of pyridine with a nickel-, cobalt-, or ruthenium-based catalyst at elevated temperatures.[86] The hydrogenation of pyridine to piperidine releases 193.8 kJ·mol−1,[87] which is slightly less than the energy of the hydrogenation of benzene (205.3 kJ·mol−1).[87]
Partially hydrogenated derivatives are obtained under milder conditions. For example, reduction with lithium aluminium hydride yields a mixture of 1,4-dihydropyridine, 1,2-dihydropyridine, and 2,5-dihydropyridine.[88] Selective synthesis of 1,4-dihydropyridine is achieved in the presence of organometallic complexes of magnesium and zinc,[89] and (Δ3,4)-tetrahydropyridine is obtained by electrochemical reduction of pyridine.[90]
Lewis basicity and coordination compounds
Pyridine is a Lewis base, donating its pair of electrons to a Lewis acid. Its Lewis base properties are discussed in the ECW model. One example is the sulfur trioxide pyridine complex (melting point 175 °C), which is a sulfation agent used to convert alcohols to sulfate esters. Pyridine-borane (C5H5NBH3, melting point 10–11 °C) is a mild reducing agent.
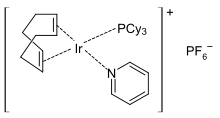
Transition metal pyridine complexes are numerous.[91][92] Typical octahedral complexes have the stoichiometry MCl2(py)4 and MCl3(py)3. Octahedral homoleptic complexes of the type M(py)6+ are rare or tend to dissociate pyridine. Numerous square planar complexes are known, such as Crabtree's catalyst.[93] The pyridine ligand replaced during the reaction is restored after its completion.
The η6 coordination mode, as occurs in η6 benzene complexes, is observed only in sterically encumbered derivatives that block the nitrogen center.[18]
Applications
Pesticides
The main use of pyridine is as a precursor to the herbicides paraquat and diquat.[2] The first synthesis step of insecticide chlorpyrifos consists of the chlorination of pyridine. Pyridine is also the starting compound for the preparation of pyrithione-based fungicides.[23] Cetylpyridinium and laurylpyridinium, which can be produced from pyridine with a Zincke reaction, are used as antiseptic in oral and dental care products.[6] Pyridine is easily attacked by alkylating agents to give N-alkylpyridinium salts. One example is cetylpyridinium chloride.
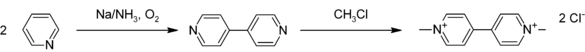
Solvent
Pyridine is used as a polar, basic, low-reactive solvent, for example in Knoevenagel condensations.[23] It is especially suitable for the dehalogenation, where it acts as the base of the elimination reaction and bonds the resulting hydrogen halide to form a pyridinium salt. In esterifications and acylations, pyridine activates the carboxylic acid halides or anhydrides. Even more active in these reactions are the pyridine derivatives 4-dimethylaminopyridine (DMAP) and 4-(1-pyrrolidinyl) pyridine. Pyridine is also used as a base in condensation reactions.[95]

It is also used in the textile industry to improve network capacity of cotton.[6]
Specialty reagents based on pyridine
As a base, pyridine can be used as the Karl Fischer reagent, but it is usually replaced by alternatives with a more pleasant odor, such as imidazole.[96]
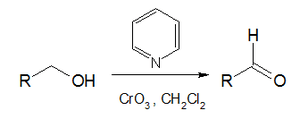
Pyridinium chlorochromate, pyridinium dichromate, and the Collins reagent (the complex of chromium(VI) oxide are used for the oxidation of alcohols.[97]
Hazards
Pyridine is added to ethanol to make it unsuitable for drinking.[6] In low doses, pyridine is added to foods to give them a bitter flavor, and such usage was approved by the US Food and Drug Administration[23] and is still considered safe by the agency even though external lobbying forced it to ban pyridine's use as a synthetic flavor in 2018.[98][99] The detection threshold for pyridine in solutions is about 1–3 mmol·L−1 (79–237 mg·L−1).[100]
Pyridine has a flash point of 17 °C and is, therefore, highly flammable. Its ignition temperature is 550 °C, and mixtures of 1.7–10.6 vol% of pyridine with air are explosive. The thermal modification of pyridine starts above 490 °C, resulting in bipyridine (mainly 2,2′-bipyridine and to a lesser extent 2,3′-bipyridine and 2,4′-bipyridine), nitrogen oxides, and carbon monoxide.[13] Pyridine easily dissolves in water and harms both animals and plants in aquatic systems.[101] The permitted maximum allowable concentration of pyridine was 15–30 parts per million (ppm, or 15–30 mg·m−3 in air) in most countries in the 1990s,[23] but was reduced to 5 ppm in the 2000s.[102] For comparison, indoor air contaminated with tobacco smoke may contain up to 16 µg·m−3 of pyridine, and one cigarette contains 21–32 µg.[23]
Health issues
Pyridine is harmful if inhaled, swallowed or absorbed through the skin.[103] Effects of acute pyridine intoxication include dizziness, headache, lack of coordination, nausea, salivation, and loss of appetite. They may progress into abdominal pain, pulmonary congestion and unconsciousness.[104] One person died after accidental ingestion of half a cup of pyridine.[23] The lowest known lethal dose (LDLo) for the ingestion of pyridine in humans is 500 mg·kg−1. In high doses, pyridine has a narcotic effect and its vapor concentrations of above 3600 ppm pose a health risk.[2] The oral LD50 in rats is 891 mg·kg−1. Pyridine is flammable.
Evaluations as a possible carcinogenic agent showed that there is inadequate evidence in humans for the carcinogenicity of pyridine, although there is limited evidence of carcinogenic effects on animals.[104] Available data indicate that "exposure to pyridine in drinking-water led to reduction of sperm motility at all dose levels in mice and increased estrous cycle length at the highest dose level in rats".[104]
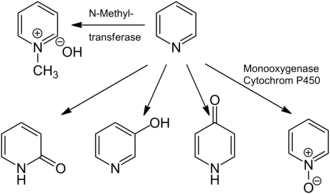
Pyridine might also have minor neurotoxic, genotoxic, and clastogenic effects.[13][23][105] Exposure to pyridine would normally lead to its inhalation and absorption in the lungs and gastrointestinal tract, where it either remains unchanged or is metabolized. The major products of pyridine metabolism are N-methylpyridiniumhydroxide, which are formed by N-methyltransferases (e.g., pyridine N-methyltransferase), as well as pyridine N-oxide, and 2-, 3-, and 4-hydroxypyridine, which are generated by the action of monooxygenase. In humans, pyridine is metabolized only into N-methylpyridiniumhydroxide.[13][105] Pyridine is readily degraded by bacteria to ammonia and carbon dioxide.[106] The unsubstituted pyridine ring degrades more rapidly than picoline, lutidine, chloropyridine, or aminopyridines,[107] and a number of pyridine degraders have been shown to overproduce riboflavin in the presence of pyridine.[108] Ionizable N-heterocyclic compounds, including pyridine, interact with environmental surfaces (such as soils and sediments) via multiple pH-dependent mechanisms, including partitioning to soil organic matter, cation exchange, and surface complexation.[109] Such adsorption to surfaces reduces bioavailability of pyridines for microbial degraders and other organisms, thus slowing degradation rates and reducing ecotoxicity.[110]
Minor amounts of pyridine are released into environment from some industrial processes such as steel manufacture,[111] processing of oil shale, coal gasification, coking plants and incinerators.[23] The atmosphere at oil shale processing plants can contain pyridine concentrations of up to 13 µg·m−3,[112] and 53 µg·m−3 levels were measured in the groundwater in the vicinity of a coal gasification plant.[113] According to a study by the US National Institute for Occupational Safety and Health, about 43,000 Americans work in contact with pyridine.[114]
Nomenclature
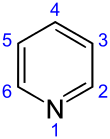
The systematic name of pyridine, within the Hantzsch–Widman nomenclature recommended by the IUPAC, is azinine. However, systematic names for simple compounds are used very rarely; instead, heterocyclic nomenclature follows historically established common names. IUPAC discourages the use of azinine/azine in favor of pyridine.[115] The numbering of the ring atoms in pyridine starts at the nitrogen (see infobox). An allocation of positions by letter of the Greek alphabet (α-γ) and the substitution pattern nomenclature common for homoaromatic systems (ortho, meta, para) are used sometimes. Here α (ortho), β (meta), and γ (para) refer to the 2, 3, and 4 position, respectively. The systematic name for the pyridine derivatives is pyridinyl, wherein the position of the substituted atom is preceded by a number. However, the historical name pyridyl is encouraged by the IUPAC and used instead of the systematic name.[116] The cationic derivative formed by the addition of an electrophile to the nitrogen atom is called pyridinium.
See also
- 6-membered aromatic rings with one carbon replaced by another group: borabenzene, benzene, silabenzene, germabenzene, stannabenzene, phosphorine, arsabenzene, pyrylium salt
- 6-membered rings with two nitrogen atoms: diazines
- 6-membered rings with three nitrogen atoms: triazines
- 6-membered rings with four nitrogen atoms: tetrazines
- 6-membered rings with five nitrogen atoms: pentazine
- 6-membered rings with six nitrogen atoms: hexazine
References
- Nomenclature of Organic Chemistry : IUPAC Recommendations and Preferred Names 2013 (Blue Book). Cambridge: The Royal Society of Chemistry. 2014. p. 141. doi:10.1039/9781849733069-FP001. ISBN 978-0-85404-182-4.
- Shimizu, S.; Watanabe, N.; Kataoka, T.; Shoji, T.; Abe, N.; Morishita, S.; Ichimura, H. "Pyridine and Pyridine Derivatives". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a22_399.
- NIOSH Pocket Guide to Chemical Hazards. "#0541". National Institute for Occupational Safety and Health (NIOSH).
- Lide, p. 3–474
- "Pyridine - CAS#:110-86-1". ChemSrc. 8 January 2020.
- RÖMPP Online – Version 3.5. Thieme Chemistry. Stuttgart: Georg Thieme. 2009.
- "Pyridine MSDS". fishersci.com. Fisher.
- "Pyridine". Immediately Dangerous to Life and Health Concentrations (IDLH). National Institute for Occupational Safety and Health (NIOSH).
- Lide, p. 3-673
- Lide, p. 5-28
- Lide, p. 6-211
- Lide, p. 6-221
- Record of Pyridine in the GESTIS Substance Database of the Institute for Occupational Safety and Health
- Majer, V.; Svoboda, V. (1985). Enthalpies of Vaporization of Organic Compounds: A Critical Review and Data Compilation. Oxford: Blackwell Scientific Publications. ISBN 0-632-01529-2.
- Domalski, Eugene S.; Hearing, Elizabeth D. (1996). "Heat Capacities and Entropies of Organic Compounds in the Condensed Phase". Journal of Physical and Chemical Reference Data. 25 (1): 1. Bibcode:1996JPCRD..25....1D. doi:10.1063/1.555985.
- Lide, p. 6-67
- McCullough, J. P.; Douslin, D. R.; Messerly, J. F.; Hossenlopp, I. A.; Kincheloe, T. C.; Waddington, Guy (1957). "Pyridine: Experimental and Calculated Chemical Thermodynamic Properties between 0 and 1500 °K.; a Revised Vibrational Assignment". Journal of the American Chemical Society. 79 (16): 4289. doi:10.1021/ja01573a014.
- Elschenbroich, C. (2008). Organometallchemie (6th ed.). Vieweg & Teubner. pp. 524–525. ISBN 978-3-8351-0167-8.
- Cox, E. (1958). "Crystal Structure of Benzene". Reviews of Modern Physics. 30 (1): 159–162. Bibcode:1958RvMP...30..159C. doi:10.1103/RevModPhys.30.159.
- Mootz, D. (1981). "Crystal structures of pyridine and pyridine trihydrate". The Journal of Chemical Physics. 75 (3): 1517–1522. Bibcode:1981JChPh..75.1517M. doi:10.1063/1.442204.
- Joule, p. 14
- Joule, p. 16
- Pyridine (PDF). IARC Monographs 77. Washington DC: OSHA. 1985.
- Joule, pp. 125–141
- Davies, D. T. (1992). Aromatic Heterocyclic Chemistry. Oxford University Press. ISBN 0-19-855660-8.
- Milcent, R.; Chau, F. (2002). Chimie organique hétérocyclique: Structures fondamentales. EDP Sciences. pp. 241–282. ISBN 2-86883-583-X.
- Krygowski, T. M.; Szatyowicz, H.; Zachara, J. E. (2005). "How H-bonding Modifies Molecular Structure and π-Electron Delocalization in the Ring of Pyridine/Pyridinium Derivatives Involved in H-Bond Complexation". J. Org. Chem. 70 (22): 8859–8865. doi:10.1021/jo051354h. PMID 16238319.
- Joule, p. 7
- Weissberger, A.; Klingberg, A.; Barnes, R. A.; Brody, F.; Ruby, P.R. (1960). Pyridine and its Derivatives. 1. New York: Interscience.
- Anderson, Thomas (1849). "On the constitution and properties of picoline, a new organic base from coal-tar". Transactions of the Royal Society of Edinburgh. 16 (2): 123–136. doi:10.1017/S0080456800024984.
- Anderson, T. (1849). "Producte der trocknen Destillation thierischer Materien" [Products of the dry distillation of animal matter]. Annalen der Chemie und Pharmacie (in German). 70: 32–38. doi:10.1002/jlac.18490700105.
- Anderson, Thomas (1851). "On the products of the destructive distillation of animal substances. Part II". Transactions of the Royal Society of Edinburgh. 20 (2): 247–260. doi:10.1017/S0080456800033160. From p. 253: "Pyridine. The first of these bases, to which I give the name of pyridine, … "
- Anderson, T. (1851). "Ueber die Producte der trocknen Destillation thierischer Materien" [On the products of dry distillation of animal matter]. Annalen der Chemie und Pharmacie (in German). 80: 44–65. doi:10.1002/jlac.18510800104.
- Koerner, W. (1869). "Synthèse d'une base isomère à la toluidine" [Synthesis of a base [that is] isomeric to toluidine]. Giornale di Scienze Naturali ed Economiche (Journal of Natural Science and Economics (Palermo, Italy)) (in French). 5: 111–114.
- Dewar, James (27 January 1871). "On the oxidation products of picoline". Chemical News. 23: 38–41.
- Rocke, Alan J. (1988). "Koerner, Dewar and the Structure of Pyridine". Bulletin for the History of Chemistry. 2: 4.

- Ladenburg, Albert. Lectures on the history of the development of chemistry since the time of Lavoisier. (PDF). pp. 283–287.
- Bansal, Raj K. (1999). Heterocyclic Chemistry. p. 216. ISBN 81-224-1212-2.
- See:
- Ladenburg, A. (1884). "Synthese des Piperidins" [Synthesis of piperidine]. Berichte der Deutschen Chemischen Gesellschaft (in German). 17: 156. doi:10.1002/cber.18840170143.
- Ladenburg, A. (1884). "Synthese des Piperidins und seiner Homologen" [Synthesis of piperidine and its homologues]. Berichte der Deutschen Chemischen Gesellschaft (in German). 17: 388–391. doi:10.1002/cber.188401701110.
- Ramsay, William (1876). "On picoline and its derivatives". Philosophical Magazine. 5th series. 2 (11): 269–281. doi:10.1080/14786447608639105.
- "A. Henninger, aus Paris. 12. April 1877". Berichte der Deutschen Chemischen Gesellschaft (Correspondence). 10: 727–737. 1877. doi:10.1002/cber.187701001202.
- Hantzsch, A. (1881). "Condensationsprodukte aus Aldehydammoniak und ketonartigen Verbindungen" [Condensation products from aldehyde ammonia and ketone-type compounds]. Berichte der Deutschen Chemischen Gesellschaft. 14 (2): 1637–1638. doi:10.1002/cber.18810140214.
- Knoevenagel, E.; Fries, A. (1898). "Synthesen in der Pyridinreihe. Ueber eine Erweiterung der Hantzsch'schen Dihydropyridinsynthese" [Syntheses in the pyridine series. On an extension of the Hantzsch dihydropyridine synthesis]. Berichte der Deutschen Chemischen Gesellschaft. 31: 761–767. doi:10.1002/cber.189803101157.
- Chichibabin, A. E. (1924). "Über Kondensation der Aldehyde mit Ammoniak zu Pyridinebasen" [On condensation of aldehydes with ammonia to make pyridines]. Journal für Praktische Chemie. 107: 122. doi:10.1002/prac.19241070110.
- Burdock, G. A., ed. (1995). Fenaroli's Handbook of Flavor Ingredients. 2 (3rd ed.). Boca Raton: CRC Press. ISBN 0-8493-2710-5.
- Täufel, A.; Ternes, W.; Tunger, L.; Zobel, M. (2005). Lebensmittel-Lexikon (4th ed.). Behr. p. 450. ISBN 3-89947-165-2.
- Tang, Jian; Jin, Qi Zhang; Shen, Guo Hui; Ho, Chi Tang; Chang, Stephen S. (1983). "Isolation and identification of volatile compounds from fried chicken". Journal of Agricultural and Food Chemistry. 31 (6): 1287. doi:10.1021/jf00120a035.
- Shibamoto, Takayuki; Kamiya, Yoko; Mihara, Satoru (1981). "Isolation and identification of volatile compounds in cooked meat: sukiyaki". Journal of Agricultural and Food Chemistry. 29: 57–63. doi:10.1021/jf00103a015.
- Aeschbacher, HU; Wolleb, U; Löliger, J; Spadone, JC; Liardon, R (1989). "Contribution of coffee aroma constituents to the mutagenicity of coffee". Food and Chemical Toxicology. 27 (4): 227–232. doi:10.1016/0278-6915(89)90160-9. PMID 2659457.
- Buttery, Ron G.; Seifert, Richard M.; Guadagni, Dante G.; Ling, Louisa C. (1971). "Characterization of Volatile Pyrazine and Pyridine Components of Potato Chips". Journal of Agricultural and Food Chemistry. Washington, DC: ACS. 19 (5): 969–971. doi:10.1021/jf60177a020.
- Ho, Chi Tang; Lee, Ken N.; Jin, Qi Zhang (1983). "Isolation and identification of volatile flavor compounds in fried bacon". Journal of Agricultural and Food Chemistry. 31 (2): 336. doi:10.1021/jf00116a038.
- Dumont, Jean Pierre; Adda, Jacques (1978). "Occurrence of sesquiterpene in mountain cheese volatiles". Journal of Agricultural and Food Chemistry. 26 (2): 364. doi:10.1021/jf60216a037.
- Labows, John N., Jr.; Warren, Craig B. (1981). "Odorants as Chemical Messengers". In Moskowitz, Howard R. (ed.). Odor Quality and Chemical Structure. Washington, DC: American Chemical Society. pp. 195–210. doi:10.1021/bk-1981-0148.fw001. ISBN 9780841206076.
- Vitzthum, Otto G.; Werkhoff, Peter; Hubert, Peter (1975). "New volatile constituents of black tea flavor". Journal of Agricultural and Food Chemistry. 23 (5): 999. doi:10.1021/jf60201a032.
- Kostelc, J. G.; Preti, G.; Nelson, P. R.; Brauner, L.; Baehni, P. (1984). "Oral Odors in Early Experimental Gingivitis". Journal of Periodontology Research. 19 (3): 303–312. doi:10.1111/j.1600-0765.1984.tb00821.x. PMID 6235346.
- Täufel, A.; Ternes, W.; Tunger, L.; Zobel, M. (2005). Lebensmittel-Lexikon (4th ed.). Behr. p. 226. ISBN 3-89947-165-2.
- Gossauer, A. (2006). Struktur und Reaktivität der Biomoleküle. Weinheim: Wiley-VCH. p. 488. ISBN 3-906390-29-2.
- "Pyridine's Development in China". AgroChemEx. 11 May 2010. Archived from the original on 20 September 2018. Retrieved 7 January 2011.
- "About Vertellus". vertellus.com. Archived from the original on 18 September 2012. Retrieved 7 January 2011.
- Frank, R. L.; Seven, R. P. (1949). "Pyridines. IV. A Study of the Chichibabin Synthesis". Journal of the American Chemical Society. 71 (8): 2629–2635. doi:10.1021/ja01176a008.
- DE patent 1917037, ICI, issued 1968
- JP patent 7039545, Nippon Kayaku, issued 1967
- BE patent 758201, Koei Chemicals, issued 1969
- Mensch, F. (1969). Erdöl Kohle Erdgas Petrochemie. 2: 67–71
- Behr, A. (2008). Angewandte homogene Katalyse. Weinheim: Wiley-VCH. p. 722. ISBN 978-3-527-31666-3.
- Kroehnke, Fritz (1976). "The Specific Synthesis of Pyridines and Oligopyridines". Synthesis. 1976 (1): 1–24. doi:10.1055/s-1976-23941..
- Skell, P. S.; Sandler, R. S. (1958). "Reactions of 1,1-Dihalocyclopropanes with Electrophilic Reagents. Synthetic Route for Inserting a Carbon Atom Between the Atoms of a Double Bond". Journal of the American Chemical Society. 80 (8): 2024. doi:10.1021/ja01541a070.
- Jones, R. L.; Rees, C. W. (1969). "Mechanism of heterocyclic ring expansions. Part III. Reaction of pyrroles with dichlorocarbene". Journal of the Chemical Society C: Organic (18): 2249. doi:10.1039/J39690002249.
- Gambacorta, A.; Nicoletti, R.; Cerrini, S.; Fedeli, W.; Gavuzzo, E. (1978). "Trapping and structure determination of an intermediate in the reaction between 2-methyl-5-t-butylpyrrole and dichlorocarbene". Tetrahedron Letters. 19 (27): 2439. doi:10.1016/S0040-4039(01)94795-1.
- Gattermann, L.; Skita, A. (1916). "Eine Synthese von Pyridin-Derivaten" [A synthesis of pyridine derivatives]. Chemische Berichte. 49 (1): 494–501. doi:10.1002/cber.19160490155.
- "Gattermann–Skita". Institute of Chemistry, Skopje. Archived from the original on 16 June 2006.
- Scott (1967). "A method for the degradation of radioactive nicotinic acid". Biochemical Journal. 102 (1): 87–93. doi:10.1042/bj1020087. PMC 1270213. PMID 6030305.
- Tarr, J. B.; Arditti, J. (1982). "Niacin Biosynthesis in Seedlings of Zea mays". Plant Physiology. 69 (3): 553–556. doi:10.1104/pp.69.3.553. PMC 426252. PMID 16662247.
- Campaigne, E. (1986). "Adrien Albert and the Rationalization of Heterocyclic chemistry". J. Chem. Educ. 63 (10): 860. Bibcode:1986JChEd..63..860C. doi:10.1021/ed063p860.
- Bakke, Jan M.; Hegbom, Ingrid (1994). "Dinitrogen Pentoxide-Sulfur Dioxide, a New nitrate ion system". Acta Chemica Scandinavica. 48: 181–182. doi:10.3891/acta.chem.scand.48-0181.
- Ono, Noboru; Murashima, Takashi; Nishi, Keiji; Nakamoto, Ken-Ichi; Kato, Atsushi; Tamai, Ryuji; Uno, Hidemitsu (2002). "Preparation of Novel Heteroisoindoles from nitropyridines and Nitropyridones". Heterocycles. 58: 301. doi:10.3987/COM-02-S(M)22.
- Duffy, Joseph L.; Laali, Kenneth K. (1991). "Aprotic Nitration (NO+
2BF−
4) of 2-Halo- and 2,6-Dihalopyridines and Transfer-Nitration Chemistry of Their N-Nitropyridinium Cations". The Journal of Organic Chemistry. 56 (9): 3006. doi:10.1021/jo00009a015. - Joule, p. 126
- Möller, Ernst Friedrich; Birkofer, Leonhard (1942). "Konstitutionsspezifität der Nicotinsäure als Wuchsstoff bei Proteus vulgaris und Streptobacterium plantarum" [Constitutional specificity of nicotinic acid as a growth factor in Proteus vulgaris and Streptobacterium plantarum]. Berichte der Deutschen Chemischen Gesellschaft (A and B Series). 75 (9): 1108. doi:10.1002/cber.19420750912.
- Mosher, H. S.; Turner, L.; Carlsmith, A. (1953). "Pyridine-N-oxide". Org. Synth. 33: 79. doi:10.15227/orgsyn.033.0079.
- Louis-Charles Campeau and Keith Fagnou (2011). "Synthesis Of 2-aryl Pyridines By Palladium-catalyzed Direct Arylation Of Pyridine N-oxides". Org. Synth. 88: 22. doi:10.15227/orgsyn.088.0022.CS1 maint: uses authors parameter (link)
- Joule, p. 133
- Shreve, R. Norris; Riechers, E. H.; Rubenkoenig, Harry; Goodman, A. H. (1940). "Amination in the Heterocyclic Series by Sodium amide". Industrial & Engineering Chemistry. 32 (2): 173. doi:10.1021/ie50362a008.
- Badger, G; Sasse, W (1963). "The Action of Metal Catalysts on Pyridines". Advances in Heterocyclic Chemistry. Advances in Heterocyclic Chemistry. 2. p. 179. doi:10.1016/S0065-2725(08)60749-7. ISBN 9780120206025. PMID 14279523.
- Sasse, W. H. F. (1966). "2,2′-bipyridine" (PDF). Organic Syntheses. 46: 5–8. doi:10.1002/0471264180.os046.02. ISBN 0471264229. Archived from the original (PDF) on 21 January 2012.
- Eller, K.; Henkes, E.; Rossbacher, R.; Hoke, H. "Amines, aliphatic". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH.
- Cox, J. D.; Pilcher, G. (1970). Thermochemistry of Organic and Organometallic Compounds. New York: Academic Press. pp. 1–636. ISBN 0-12-194350-X.
- Tanner, Dennis D.; Yang, Chi Ming (1993). "On the structure and mechanism of formation of the Lansbury reagent, lithium tetrakis(N-dihydropyridyl) aluminate". The Journal of Organic Chemistry. 58 (7): 1840. doi:10.1021/jo00059a041.
- De Koning, A.; Budzelaar, P. H. M.; Boersma, J.; Van Der Kerk, G. J. M. (1980). "Specific and selective reduction of aromatic nitrogen heterocycles with the bis-pyridine complexes of bis(1,4-dihydro-1-pyridyl)zinc and bis(1,4-dihydro-1-pyridyl)magnesium". Journal of Organometallic Chemistry. 199 (2): 153. doi:10.1016/S0022-328X(00)83849-8.
- Ferles, M. (1959). Collection of Czechoslovak Chemical Communications. 24: 1029–1033.
- Nakamoto, K. (1997). Infrared and Raman spectra of Inorganic and Coordination compounds. Part A (5th ed.). Wiley. ISBN 0-471-16394-5.
- Nakamoto, K. (31 July 1997). Infrared and Raman spectra of Inorganic and Coordination compounds. Part B (5th ed.). p. 24. ISBN 0-471-16392-9.
- Crabtree, Robert (1979). "Iridium compounds in catalysis". Accounts of Chemical Research. 12 (9): 331–337. doi:10.1021/ar50141a005.
- "Environmental and health criteria for paraquat and diquat". Geneva: World Health Organization. 1984.
- Sherman, A. R. (2004). "Pyridine". In Paquette, L. (ed.). Encyclopedia of Reagents for Organic Synthesis. e-EROS (Encyclopedia of Reagents for Organic Synthesis). New York: J. Wiley & Sons. doi:10.1002/047084289X.rp280. ISBN 0471936235.
- "Wasserbestimmung mit Karl-Fischer-Titration" [Water analysis with the Karl Fischer titration] (PDF). Jena University. Archived from the original (PDF) on 19 July 2011.
- Tojo, G.; Fernandez, M. (2006). Oxidation of alcohols to aldehydes and ketones: a guide to current common practice. New York: Springer. pp. 28, 29, 86. ISBN 0-387-23607-4.
- 83 FR 50490
- "FDA Removes 7 Synthetic Flavoring Substances from Food Additives List". 5 October 2018. Retrieved 8 October 2018.
- Täufel, A.; Ternes, W.; Tunger, L.; Zobel, M. (2005). Lebensmittel-Lexikon (4th ed.). Behr. p. 218. ISBN 3-89947-165-2.
- "Database of the (EPA)". U.S. Environmental Protection Agency.
- "Pyridine MSDS" (PDF). Alfa Aesar. Retrieved 3 June 2010.
- Aylward, G (2008). SI Chemical Data (6th ed.). ISBN 978-0-470-81638-7.
- International Agency for Research on Cancer (IARC) (22 August 2000). "Pyridine Summary & Evaluation". IARC Summaries & Evaluations. IPCS INCHEM. Retrieved 17 January 2007.
- Bonnard, N.; Brondeau, M. T.; Miraval, S.; Pillière, F.; Protois, J. C.; Schneider, O. "Pyridine" (PDF). Fiche Toxicologique (in French). INRS.
- Sims, G. K.; O'Loughlin, E. J. (1989). "Degradation of pyridines in the environment". CRC Critical Reviews in Environmental Control. 19 (4): 309–340. doi:10.1080/10643388909388372.
- Sims, G. K.; Sommers, L.E. (1986). "Biodegradation of pyridine derivatives in soil suspensions". Environmental Toxicology and Chemistry. 5 (6): 503–509. doi:10.1002/etc.5620050601.
- Sims, G. K.; O'Loughlin, E.J. (1992). "Riboflavin production during growth of Micrococcus luteus on pyridine". Applied and Environmental Microbiology. 58 (10): 3423–3425. doi:10.1128/AEM.58.10.3423-3425.1992. PMC 183117. PMID 16348793.
- Bi, E.; Schmidt, T. C.; Haderlein, S. B. (2006). "Sorption of heterocyclic organic compounds to reference soils: column studies for process identification". Environ Sci Technol. 40 (19): 5962–5970. Bibcode:2006EnST...40.5962B. doi:10.1021/es060470e. PMID 17051786.
- O'Loughlin, E. J; Traina, S. J.; Sims, G. K. (2000). "Effects of sorption on the biodegradation of 2-methylpyridine in aqueous suspensions of reference clay minerals". Environmental Toxicology and Chemistry. 19 (9): 2168–2174. doi:10.1002/etc.5620190904.
- Junk, G. A.; Ford, C. S. (1980). "A review of organic emissions from selected combustion processes". Chemosphere. 9 (4): 187. Bibcode:1980Chmsp...9..187J. doi:10.1016/0045-6535(80)90079-X.
- Hawthorne, Steven B.; Sievers, Robert E. (1984). "Emissions of organic air pollutants from shale oil wastewaters". Environmental Science & Technology. 18 (6): 483. Bibcode:1984EnST...18..483H. doi:10.1021/es00124a016. PMID 22247953.
- Stuermer, Daniel H.; Ng, Douglas J.; Morris, Clarence J. (1982). "Organic contaminants in groundwater near to underground coal gasification site in northeastern Wyoming". Environmental Science & Technology. 16 (9): 582–7. Bibcode:1982EnST...16..582S. doi:10.1021/es00103a009. PMID 22284199.
- National Occupational Exposure Survey 1981–83. Cincinnati, OH: Department of Health and Human Services, Public Health Service, Centers for Disease Control, National Institute for Occuptional Safety and Health.
- Powell, W. H. (1983). "Revision of the extended Hantzsch-Widman system of nomenclature for hetero mono-cycles" (PDF). Pure and Applied Chemistry. 55 (2): 409–416. doi:10.1351/pac198855020409.
- Hellwinkel, D. (1998). Die systematische Nomenklatur der Organischen Chemie (4th ed.). Berlin: Springer. p. 45. ISBN 3-540-63221-2.
Bibliography
- Joule, J. A.; Mills, K. (2010). Heterocyclic Chemistry (5th ed.). Chichester: Blackwell Publishing. ISBN 978-1-4051-3300-5.
- Lide, D. R., ed. (2009). Handbook of Chemistry and Physics (90th ed.). Boca Raton: CRC Press. ISBN 978-1-4200-9084-0.
External links
| Wikimedia Commons has media related to Pyridine. |
- Synthesis and propierties of pyridines at chemsynthesis.com
- International Chemical Safety Card 0323
- NIOSH Pocket Guide to Chemical Hazards
- Synthesis of pyridines (overview of recent methods)
| Authority control |
|
|---|