Kröhnke pyridine synthesis
The Kröhnke pyridine synthesis is reaction in organic synthesis between α-pyridinium methyl ketone salts and α, β-unsaturated carbonyl compounds used to generate highly functionalized pyridines. Pyridines occur widely in natural and synthetic products, so there is wide interest in routes for their synthesis. The method is named after Dr. Fritz Kröhnke.
| Kröhnke pyridine synthesis | |
|---|---|
| Named after | Fritz Kröhnke |
| Reaction type | Ring forming reaction |
| Identifiers | |
| RSC ontology ID | RXNO:0000420 |
Reaction development
Discovery
In his work at the University of Giessen, Kröhnke observed condensation of α-pyridinium methyl ketone salts 1 with α,β-unsaturated carbonyl compounds 2 via a Michael reaction when treated with ammonium acetate to give 2,4,6-trisubstituted pyridines in high yields under mild reaction conditions.[1] The proposed intermediates, 1, 5-dicarbonyl compound 3, have not been isolated.[2] Since its discovery, the Kröhnke synthesis has enjoyed broad applicability to the preparation of di-,tri- and tetrapyridine derivatives, demonstrating a number of advantages over related reactions such as the Hantzsch pyridine synthesis.
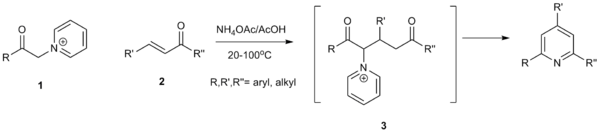
Mechanism
The mechanism of the Kröhnke pyridine synthesis begins with enolization of α-pyridinium methyl ketone 4 followed by 1,4-addition to the α, β-unsaturated ketone 5 to form the Michael adduct 6, which immediately tautomerizes to the 1,5-dicarbonyl 7. Addition of ammonia to 7 followed by dehydration via 8 generates the imine intermediate 9.,[3][4] The imine intermediate is then deprotonated to enamine 10 and cyclizes with the carbonyl to generate intermediate 11. The pyridinium cation is then eliminated to form hydroxy-dienamine 12. Aromatization of 12 via subsequent loss of water generates the desired pyridine heterocycle 13.

Reagent synthesis and reaction conditions
The starting materials for the Kröhnke synthesis are often trivial to prepare, lending to the convenience and broad scope of the method. Preparation of the α-pyridinium methyl ketone salts can be easily achieved by treatment of the corresponding α-bromo methyl ketone with pyridine. The α,β-unsaturated ketones are often available commercially or can be prepared using a number of known methods. Additionally, Mannich bases can also be utilized as the Michael acceptor for the scheme, further diversifying the scope of starting materials that can be incorporated into the Kröhnke scheme.[5]
The reaction conditions for the Kröhnke synthesis are generally facile and the reactions often proceed in high yields with reaction temperatures generally not exceeding 140 °C.[6] The Kröhnke synthesis is generally performed in either glacial acetic acid or methanol, but it can also be done under aqueous conditions, and more recently under solvent-free conditions.
1,3-dicarbonyl compounds have also been shown to be viable starting materials in place of the α-pyridinium methyl ketone salts.[7] For example, treatment of 1,3-diketone 14 with base in ethanol followed by ammonium acetate, acetic acid, the corresponding enone and a Lewis acid yields 3-acyltriarylpyridines of the form 15. These acyl pyridine are attractive intermediates because they have an electrophilic handle that allows for additional functionality to be incorporated into the molecule. This allows for straightforward construction of complex polyaryl systems, an attractive method for library synthesis of drug targets containing functionalized pyridine moieties.

Advantages over other methods
The Kröhnke synthesis for making pyridines possesses a number of succinct advantages over other methods. Unlike the Hantzsch synthesis,[8] the Kröhnke method does not require oxidation to generate the desired product since the α-pyridinium methyl ketone already possesses the correct oxidation state.
Another advantage of the Kröhnke synthesis is its high atom economy. For example, the Chichibabin synthesis requires 2 equivalents of unsaturated starting material.[9] Additionally, the byproducts of the Kröhnke synthesis is water and pyridine, which allow for easy workup and purification protocols. Unlike comparable methods for pyridine synthesis, the Kröhkne synthesis benefits from being a high-yielding one pot synthesis, which ultimately allows for abbreviation of synthetic pathways and further simplifies combinatorial library cataloging.
Scope and limitations
The broad scope of the Kröhnke pyridine synthesis has made it particularly useful for the synthesis of poly aryl systems including pyridyl, thienyl, and furanyl moieties as well. The method tolerates a broad array of aryl substitiuents on both the α-pyridinium methyl ketone fragment and the α, β-unsaturated carbonyl compounds and can thus be used to generate a wide catalog of poly-aryl systems. Additionally, electron-withdrawing groups and electron-donating groups on the incoming aryl substituents are both well tolerated. The Kröhnke synthesis can also employ alkyl and vinyl substituents giving moderated to good yields as well.[10] Due to its broad scope, the Kröhnke method has seen wide applicability to for the synthesis of bipyridines (16), terpyridines (17), quaterpyridines (18) and even up to septipyridines (19) as shown below.[11]

Variations and combinatorial studies
The Kröhnke method is featured in a solvent-free synthesis of triarylpyridines that proceeds via a homo-coupling of two diaryl substituted α, β-unsaturated carbonyl compounds.[12] This strategy offers a facile means for preparation of pyridnyl aryl systems that are important fragments of many useful drug scaffolds.

In 1992, Robinson and co-workers developed a similar pyridine synthesis using enamino nitriles as one of the three-carbon fragments in place of an α-pyridinium methyl ketone.[13] This improvement increases the reactivity of the system and allows for formation of fully substituted pyridines whereas use of an α-pyridinium methyl ketone requires that the 3- or 5- position on the resulting pyridine be unsubstituted. Kröhnke condensation of enamino nitrile 20 with enone 21 yielded fused pyridine 22.

The mechanism of this Kröhnke-type reaction likely proceeds via a vinylogous cyanamide 23 which undergoes elimination of hydrocyanic acid, deprotonation to form enamine 24 and cyclization to form intermediate 25, which is then dehydrated to form the desired pyridine product.

A clean one-pot Kröhnke method in aqueous media generates 4’-aryl-2,2’:6’, 2’’-terpyridines.[14] Reaction of aryl aldehyde 26 with two equivalents of 2-acetylpyridine (27) yielded terpyridines of the form 28.

In addition to variations on the original method, a number of combinatorial studies using the Kröhnke synthesis and its variations have been employed to synthesize vast libraries of highly functionalized pyridines. Janda and co-workers utilized the general Kröhnke reaction scheme to generate a 220 compound library.[15] Various methyl ketones 29 and aldehydes 30 were coupled via aldol condensation to give enones of the form 31. These compounds were then reacted with various α-pyridinium methyl ketones 32 to give the desired tri-substituted pyridine 33.

In 2009, Tu and coworkers developed a 3 fragment, one-pot combinatorial strategy for developing 3-cyanoterpyridines 34and 1-amino-2-acylterpyridines 35.[16] These combinatorial variations of the Kröhnke reaction provide an efficient synthetic strategy to poly arylpyridine scaffolds. This methodology would also be advantageous for biological assays and screening experiments.

Synthetic applications to ligands and biologically active molecules
The Kröhnke methodology has also been utilized to generate a number of interesting metal-binding ligands since polypyridyl complexes such as bipyridine (bipy) have been used extensively as ligands. The Kröhnke synthesis was used to prepare a family of tetrahydroquinoline-based N, S-type ligands.[17] 2-thiophenylacetophenone (36) was reacted with iodine gas and pyridine in quantitative yield to generate acylmethylpyridinium iodide 37. Reaction with a chiral cyclic α, β-unsaturated ketone derived from 2-(+)-carene yielded the desired N, S-type ligand 38.

Novel, chiral P, N-ligands have been prepared using the Kröhnke method.[18] α-pyridinium acyl ketone salt 39 was cyclized with pinocarvone derivative 40 to generate pyridine 41. The benzylic position of 41 was methylated and subsequent SnAr reaction with potassium diphenylphosphide to generate ligand 42.

The Kröhnke reaction has also enjoyed applicability to the synthesis of a number of biologically active compounds in addition to ones cataloged in combinatorial studies. Kelly and co-workers developed a route to cyclo-2,2′:4′,4′′:2′′,2′′′:4′′′,4′′′′:2′′′′,2′′′′′:4′′′′′,4-sexipyridine utilizing the Kröhnke reactions as the key macrocyclization step.[19] Polypyridine complex 43 was treated with N-Bromosuccinimide in wet tetrahydrofuran followed by pyridine to generate the acylmethylpyridinium salt 44 which can then undergo the macrocyclization under standard conditions to yield the desired product 45. The Kröhnke method in this synthesis was crucial due to the failure of other cyclization techniques such as the Glaser coupling or Ullmann coupling.

Another use of the Kröhnke pyridine synthesis was the generation of a number of 2,4,6-trisubstituted pyridines that were investigated as potential topoisomerase 1 inhibitors.[20] 2-acetylthiophene (46) was treated with iodine and pyridine to generate α-pyridinium acyl ketone 47. Reaction with Michael acceptor 48 under standard conditions yielded functionalized pyridine 49 in 60% overall yield.

Ultimately, the Kröhnke pyridine synthesis offers a facile and straightforward approach to the synthesis of a wide breadth of functionalized pyridines and poly aryl systems. The Kröhnke methodology has been applied to a number of strategies towards interesting ligands and biologically-relevant molecules. Additionally, the Kröhnke reaction and its variations offer a number of advantages than alternative methods to pyridine synthesis ranging from one-pot, organic solvent-free variations to high atom economy.
See also
- Hantzsch pyridine synthesis
- Gattermann–Skita synthesis
- Chichibabin pyridine synthesis
- Ciamician-Dennstedt rearrangement
- Bönnemann cyclization
References
- Zecher, W.; Kröhnke, F. Ber. 1961, 94, 690-697.
- Kröhnke, F.; Zecher, W. Angewandte Chemie International Edition in English 1962, Volume 1, pages 626–632. doi:10.1002/anie.196206261
- Potts, K. T.; Cipullo, M. J.; Ralli, P.; Theodoridis, G. J. Am. Chem. Soc. 1981, 103, 3584-3586.
- Kelly, T. R.; Lee, Y. J.; Mears, R. J. J. Org. Chem. 1997, 62, 2774-2781
- Kröhnke, F.; Zecher, W.; Angew. Chem. 1963, 75, 189
- Kröhnke, F. Synthesis. 1976, 1, 1-24
- Rehberg, R. W.; Kröhnke, F. Justus Liebigs Ann. Chem.1968, 91, 717
- Hantzsch, A. (1881). "Condensationprodukte aus Aldehydammoniak und Ketonartigen Verbindungen". Chemische Berichte 14 (2): 1637
- Chichibabin, A. E. J. prakt. Chem. 1924, 107, 122
- Kürti László, Barbara Czakó. Strategic Applications of Named Reactions in Organic Synthesis. Elsevier Inc.: Burlington, Massachusetts.
- Kröhnke, F.; Kröck, F. W.; Chem Ber. 1971, 104, 1645
- Adib, M.; Tahermansouri, H.; Koloogani, S. A.; Mohammadi, B.; Bijanzadej, H. R. Tetrahedron Lett.2006, 47, 5957-5960
- Robinson et. al. J. Org. Chem. 1992, 57, 7352
- Tu, S.; Jia, R.; Jiang, B.; Zhang, J.; Zhang, Y.; Yao, C.; Ji, S. Tetrahedron, 2007, 63, 381-388
- Janda, K. D.; Wirsching, P.; Fujimori, T. J. Comb. Chem.2003, 5, 625-631
- Tu, S.; Jiang, B.; Hao, W.; Wang, X.; Shi, F. J. Comb. Chem. 2009, 11, 846-850
- Chelucci, G. et al. J. Mol. Catal. A: Chemical, 2003, 191, 1-8
- Andrei V. Malkov, Marco Bella, Irena G. Stara, P. Kocovsky "Modular pyridine-type P,N-ligands derived from monoterpenes: application in asymmetric Heck addition" Tetrahedron Lett. 2001, 42, 3045-3048. doi:10.1016/S0040-4039(01)00369-0
- Kelly, T. J. Org. Chem. 1997, 62, 2774-2781
- Lee, E.-S. Med. Chem. Lett. 2004, 14, 1333-1337