Janus kinase 3
Tyrosine-protein kinase JAK3 is a tyrosine kinase enzyme that in humans is encoded by the JAK3 gene.[5][6]
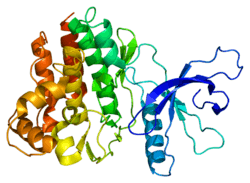


Janus kinases
Janus kinase 3 is a tyrosine kinase that belongs to the janus family of kinases. Other members of the Janus family include JAK1, JAK2 and TYK2. Janus kinases (JAKs) are relatively large kinases of approximately 1150 amino acids with apparent molecular weights of 120-130 kDa.[7] They are cytosolic tyrosine kinases that are specifically associated with cytokine receptors. Since cytokine receptor proteins lack enzymatic activity, they are dependent upon JAKs to initiate signaling upon binding of their ligands (e.g. cytokines). The cytokine receptors can be divided into five major subgroups based on their different domains and activation motifs. JAK3 is required for signaling of the type I receptors that use the common gamma chain (γc).
| Type | Subgroup | Cytokine Receptor | JAK Kinase |
|---|---|---|---|
| I | homodimeric | EPO, TPO, GH, G-CSF | JAK2 |
| uses common beta chain (CSF2RΒ) | IL-3, IL-5, GM-CSF | JAK2 | |
| uses gp130 chain | IL-6, IL-11 | JAK1, JAK2, Tyk2 | |
| uses common gamma chain (γc) | IL-2, IL-4, IL-7, IL-9, IL-15, IL-21 | JAK1, JAK3 | |
| II | IFN-α, IFN-β, IFN-γ | JAK1, JAK2, Tyk2 |
Function
As JAK3 is expressed in hematopoietic and epithelial cells, its role in cytokine signaling is thought to be more restricted than other JAKs. It is most commonly expressed in T cells and NK cells,[7] but has also been found in intestinal epithelial cells.[9][10][11] JAK3 is involved in signal transduction by receptors that employ the common gamma chain (γc) of the type I cytokine receptor family (e.g. IL-2R, IL-4R, IL-7R, IL-9R, IL-15R, and IL-21R).[12] Mutations that abrogate Janus kinase 3 function cause an autosomal SCID (severe combined immunodeficiency disease),[13] while activating Janus kinase 3 mutations lead to the development of leukemia.[14]
In addition to its well-known roles in T cells and NK cells, JAK3 has been found to mediate IL-8 stimulation in human neutrophils. IL-8 primarily functions to induce chemotaxis in neutrophils and lymphocytes, and JAK3 silencing severely inhibits IL-8-mediated chemotaxis.[15]
Intestinal epithelial cells
Jak3 interacts with actin-binding protein villin, thereby facilitating cytoskeletal remodeling and mucosal wound repair.[11] Structural determinants that regulate the interactions between Jak3 and cytoskeletal proteins of the villin / gelsolin family have also been characterized. Functional reconstitution of kinase activity by recombinant Jak3 using Jak3-wt or villin/gelsolin-wt as substrate showed that Jak3 autophosphorylation was the rate-limiting step during interactions between Jak3 and cytoskeletal proteins. Kinetic parameters showed that phosphorylated (P) Jak3 binds to P-villin with a dissociation constant (Kd) of 23 nM and a Hill's coefficient of 3.7. Pairwise binding between Jak3 mutants and villin showed that the FERM domain of Jak3 was sufficient for binding to P-villin with a Kd of 40.0 nM. However, the SH2 domain of Jak3 prevented P-villin from binding to the FERM domain of nonphosphorylated protein. The intramolecular interaction between the FERM and SH2 domains of nonphosphorylated Jak3 prevented Jak3 from binding to villin and tyrosine autophosphorylation of Jak3 at the SH2 domain decreased these intramolecular interactions and facilitated binding of the FERM domain to villin. These demonstrate the molecular mechanism of interactions between Jak3 and cytoskeletal proteins where tyrosine phosphorylation of the SH2 domain acted as an intramolecular switch for the interactions between Jak3 and cytoskeletal proteins.[9]
Sustained damage to the mucosal lining in patients with inflammatory bowel disease (IBD) facilitates translocation of intestinal microbes to submucosal immune cells leading to chronic inflammation. IL-2 plays a role in intestinal epithelial cell (IEC) homeostasis through concentration-dependent regulation of IEC proliferation and cell death. Activation by IL-2 led to tyrosine phosphorylation-dependent interactions between Jak3 and p52ShcA only at lower concentrations. Higher concentrations of IL-2 decreased the phosphorylation of Jak3, disrupted its interactions with p52ShcA, redistributed Jak3 to the nucleus, and induced apoptosis in IEC. IL-2 also induced dose-dependent downregulation of jak3-mRNA. Constitutive overexpression and mir-shRNA-mediated knockdown studies showed that expression of Jak3 was necessary for IL-2-induced proliferation of IEC. Additionally, IL-2-induced downregulation of jak3-mRNA was responsible for higher IL-2-induced apoptosis in IEC. Thus IL-2-induced mucosal homeostasis through posttranslational and transcriptional regulation of Jak3.[10]
Jak3 is also implicated in mucosal differentiation and predisposition to inflammatory bowel disease in mice model. These studies show that Jak3 is expressed in colonic mucosa of mice, and the loss of mucosal expression of Jak3 results in reduced expression of differentiation markers for the cells of both enterocytic and secretory lineages. Jak3 KO mice showed reduced expression of colonic villin, carbonic anhydrase, secretory mucin muc2, and increased basal colonic inflammation reflected by increased levels of pro-inflammatory cytokines IL-6 and IL-17A in colon along with increased colonic myeloperoxidase activity. The inflammations in KO mice were associated with shortening of colon length, reduced cecum length, decreased crypt heights, and increased severity toward dextran sulfate sodium-induced colitis. In differentiated human colonic epithelial cells, Jak3 redistributed to basolateral surfaces and interacted with adherens junction (AJ) protein β-catenin. Jak3 expression in these cells was essential for AJ localization of β-catenin and maintenance of epithelial barrier functions. Collectively, these results demonstrate the essential role of Jak3 in the colon where it facilitated mucosal differentiation by promoting the expression of differentiation markers and enhanced colonic barrier functions through AJ localization of β-catenin.[16]
Though constitutive activation of Janus kinase 3 (Jak3) leads to different cancers, the mechanism of trans-molecular regulation of Jak3 activation is only recently reported. This study showed that Jak3 auto-phosphorylation was the rate limiting step during Jak3 trans-phosphorylation of Shc where Jak3 directly phosphorylated (P) two tyrosine residues in SH-2-domain, and one tyrosine residue each in CH-1, and PID domains of Shc. Direct interactions between mutants of Jak3 and Shc showed that while FERM domain of Jak3 was sufficient for binding to Shc, CH-1 and PID domains of Shc were responsible for binding to Jak3. Functionally, Jak3 was auto-phosphorylated under IL-2 stimulation in epithelial cells. However, Shc recruited tyrosine phosphatase SHP-2 and PTP-1B to Jak3 and thereby dephosphorylate Jak3. Thus the study not only characterized Jak3 interaction with Shc, but also demonstrated the mechanism of intracellular regulation of Jak3 activation where Jak3 interactions with Shc acted as a regulator of Jak3 dephosphorylation through direct interactions of Shc with both Jak3 and tyrosine phosphatases.[17]
Chronic low-grade inflammation (CLGI) plays a key role in metabolic deterioration in the obese population. Jak3 expression and activation provide protection against development of CLGI and associated health complications. Studies in rodent model show that loss of Jak3 results in increased body weight, basal systemic CLGI, compromised glycemic homeostasis, hyperinsulinemia, and early symptoms of liver steatosis. Lack of Jak3 also results in exaggerated symptoms of metabolic syndrome by western high-fat diet. Mechanistically, it is shown that Jak3 is essential for reduced expression and activation of toll like receptors (TLRs) in murine intestinal mucosa and human intestinal epithelial cells where Jak3 interacted with and activated p85, the regulatory sub-unit of the PI3K, through tyrosine phosphorylation of adapter protein insulin receptor substrate (IRS1). These interactions resulted in activation of PI3K-Akt axis, which was essential for reduced TLR expression and TLR associated NF-κB activation. Overall, Jak3 plays an essential role in promoting mucosal tolerance through suppressed expression and limiting activation of TLRs thereby preventing intestinal and systemic CLGI and associated obesity and MetS.[18]
Compromise in adherens junctions (AJs) is associated with several chronic inflammatory diseases. Functional characterization showed that Jak3 autophosphorylation was the rate-limiting step during Jak3 trans-phosphorylation of β-catenin, where Jak3 directly phosphorylated three tyrosine residues, viz. Tyr30, Tyr64, and Tyr86 in the N-terminal domain (NTD) of β-catenin. However, prior phosphorylation of β-catenin at Tyr654 was essential for further phosphorylation of β-catenin by Jak3. Interaction studies indicated that phosphorylated Jak3 bound to phosphorylated β-catenin with a dissociation constant of 0.28 μm, and although both the kinase and FERM (Band 4.1, ezrin, radixin, and moesin) domains of Jak3 interacted with β-catenin, the NTD domain of β-catenin facilitated its interactions with Jak3. Physiologically, Jak3-mediated phosphorylation of β-catenin suppressed EGF-mediated epithelial–mesenchymal transition (EMT)and facilitated epithelial barrier functions by AJ localization of phosphorylated β-catenin through its interactions with α-catenin. Moreover, loss of Jak3-mediated phosphorylation sites in β-catenin abrogated its AJ localization and compromised epithelial barrier functions. Together, this study not only characterized Jak3 interaction with β-catenin but also demonstrated the mechanism of molecular interplay between AJ dynamics and EMT by Jak3-mediated NTD phosphorylation of β-catenin.[19]
Breast cancer resistance protein (BCRP) is a member of ATP-binding cassette (ABC) transporter proteins whose primary function is to efflux substrates bound to the plasma membrane. Impaired intestinal barrier functions play a major role in chronic low-grade inflammation (CLGI)-associated obesity, but the regulation of BCRP during obesity and its role in maintaining the intestinal barrier function during CLGI-associated obesity were unknown. Using several approaches, including efflux assays, immunoprecipitation/-blotting/-histochemistry, paracellular permeability assay, fluorescence activated cell sorting, cytokine assay, and immunofluorescence microscopy, recent studies suggest that obese individuals have compromised intestinal BCRP functions and that diet-induced obese mice recapitulate these outcomes. It was also demonstrated that the compromised BCRP functions during obesity were due to loss of Janus kinase 3 (JAK3)-mediated tyrosine phosphorylation of BCRP. Results in the studies indicated that JAK3-mediated phosphorylation of BCRP promotes its interactions with membrane-localized β-catenin essential not only for BCRP expression and surface localization, but also for the maintenance of BCRP-mediated intestinal drug efflux and barrier functions. It was observed that reduced intestinal JAK3 expression during human obesity or JAK3 knockout in mouse or siRNA-mediated β-catenin knockdown in human intestinal epithelial cells all result in significant loss of intestinal BCRP expression and compromised colonic drug efflux and barrier functions. These results uncover a mechanism of BCRP-mediated intestinal drug efflux and barrier functions and establish a role for BCRP in preventing CLGI-associated obesity both in humans and in mice. These studies have wider implications not only in our understanding of physiological and pathophysiological mechanisms of intestinal barrier functions and CLGI associated chronic inflammatory diseases but also in protein-mediated drug-efflux pharmacokinetic and pharmacodynamic characteristics of oral drug formulations.[20]
Signal transduction model
JAK3 is activated only by cytokines whose receptors contain the common gamma chain (γc) subunit: IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21. Cytokine binding induces the association of separate cytokine receptor subunits and the activation of the receptor-associated JAKs. In the absence of cytokine, JAKs lack protein tyrosine kinase activity. Once activated, the JAKs create docking sites for the STAT transcription factors by phosphorylation of specific tyrosine residues on the cytokine receptor subunits. STATs (signal transduction and activators of transcription) are members of a family of transcription factors, and they have src homology 2 (SH2) domains that allow them to bind to these phosphorylated tyrosine residues. After undergoing JAK-mediated phosphorylation, the STAT transcription factors dimerize, translocate to the nucleus, bind DNA at specific elements and induce expression of specific genes.[7] Cytokine receptors selectively activate particular JAK-STAT pathways to induce transcription of different genes. IL-2 and IL-4 activate JAK1, JAK3 and STAT5.[21]
Disease relevance
JAK3 activating mutations are found in 16% of T-cell acute lymphoblastic leukemia (T-ALL) patients.[22] In addition, oncogenic JAK3 mutations have been identified in acute megakaryoblastic leukemia, T-cell prolymphocytic leukemia, and juvenile myelomonocytic leukemia and natural killer T-cell lymphoma (NK/T-lymphoma). Most mutations are located in the pseudokinase and kinase domain of the JAK3 protein. Most JAK3 mutations are dependent on JAK1 kinase activity for their transforming capacities.[14]
Inactivating mutations of JAK3 are known causes of immune deficiency.[23] Mutations in the common gamma chain (γc) result in X-linked severe combined immunodeficiency (X-SCID). Since γc specifically associates with JAK3, mutations in JAK3 also result in SCID.[24] Deficiency of JAK3 blocks signaling of the following cytokines and their effects:[8]
- IL-2 - T cell proliferation and maintenance of peripheral tolerance
- IL-4 - differentiation of Th2 cells
- IL-7 - thymocyte development in the thymus
- IL-9 - survival signal for various hematopoietic cells
- IL-15 - NK cell development
- IL-21 - regulation of immunoglobulin class switching in B cells
Overall, JAK3 deficiency results in the phenotype of SCID characterized by T−B+NK−, which indicates the absence of T cells and NK cells.[25] Although B cells are present, they are non-functional due to defective B cell activation and impaired antibody class switching.
Since JAK3 is required for immune cell development, targeting JAK3 could be a useful strategy to generate a novel class of immunosuppressant drugs. Moreover, unlike other JAKs, JAK3 is primarily expressed in hematopoietic cells, so a highly specific JAK3 inhibitor should have precise effects on immune cells and minimal pleiotropic defects.[8] The selectivity of a JAK3 inhibitor would also have advantages over the current widely used immunosuppressant drugs, which have abundant targets and diverse side effects. A JAK3 inhibitor could be useful for treating autoimmune diseases, especially those in which a particular cytokine receptor has a direct role on disease pathogenesis. For example, signaling through the IL-15 receptor is known to be important in the development rheumatoid arthritis,[26] and the receptors for IL-4 and IL-9 play roles in the development of allergic responses.[27]
A selective JAK3 inhibitor, designated CP-690550, has been developed and shown promise in clinical trials. This drug has nanomolar potency against JAK3 and was shown to be effective in preventing transplant rejection in a nonhuman primate renal transplant model.[8] CP-690550 also demonstrated immunosuppressive activity in phase I and II clinical trials of rheumatoid arthritis, psoriasis and organ transplant rejection.[28] CP-690550 (Tofacitinib) is currently being market by Pfizer as Xeljanz for the treatment of rheumatoid arthritis.[29]
Interactions
Janus kinase 3 has been shown to interact with CD247,[30] TIAF1[31] and IL2RG.[32][33]
References
- GRCh38: Ensembl release 89: ENSG00000105639 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000031805 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Riedy MC, Dutra AS, Blake TB, Modi W, Lal BK, Davis J, Bosse A, O'Shea JJ, Johnston JA (October 1996). "Genomic sequence, organization, and chromosomal localization of human JAK3". Genomics. 37 (1): 57–61. doi:10.1006/geno.1996.0520. PMID 8921370.
- Hoffman SM, Lai KS, Tomfohrde J, Bowcock A, Gordon LA, Mohrenweiser HW (July 1997). "JAK3 maps to human chromosome 19p12 within a cluster of proto-oncogenes and transcription factors". Genomics. 43 (1): 109–11. doi:10.1006/geno.1997.4792. PMID 9226382.
- Leonard WJ, O'Shea JJ (1998). "Jaks and STATs: biological implications" (PDF). Annual Review of Immunology. 16: 293–322. doi:10.1146/annurev.immunol.16.1.293. PMID 9597132.
- O'Shea JJ, Park H, Pesu M, Borie D, Changelian P (May 2005). "New strategies for immunosuppression: interfering with cytokines by targeting the Jak/Stat pathway". Current Opinion in Rheumatology. 17 (3): 305–11. doi:10.1097/01.bor.0000160781.07174.db. PMID 15838241.
- Mishra J, Karanki SS, Kumar N (November 2012). "Identification of molecular switch regulating interactions of Janus kinase 3 with cytoskeletal proteins". The Journal of Biological Chemistry. 287 (49): 41386–91. doi:10.1074/jbc.C112.363507. PMC 3510836. PMID 23012362.
- Mishra J, Waters CM, Kumar N (March 2012). "Molecular mechanism of interleukin-2-induced mucosal homeostasis". American Journal of Physiology. Cell Physiology. 302 (5): C735-47. doi:10.1152/ajpcell.00316.2011. PMC 3311301. PMID 22116305.
- Kumar N, Mishra J, Narang VS, Waters CM (October 2007). "Janus kinase 3 regulates interleukin 2-induced mucosal wound repair through tyrosine phosphorylation of villin". The Journal of Biological Chemistry. 282 (42): 30341–5. doi:10.1074/jbc.C600319200. PMID 17537734.
- Johnston JA, Kawamura M, Kirken RA, Chen YQ, Blake TB, Shibuya K, Ortaldo JR, McVicar DW, O'Shea JJ (July 1994). "Phosphorylation and activation of the Jak-3 Janus kinase in response to interleukin-2". Nature. 370 (6485): 151–3. doi:10.1038/370151a0. PMID 8022485.
- Fujimoto M, Naka T, Nakagawa R, Kawazoe Y, Morita Y, Tateishi A, Okumura K, Narazaki M, Kishimoto T (August 2000). "Defective thymocyte development and perturbed homeostasis of T cells in STAT-induced STAT inhibitor-1/suppressors of cytokine signaling-1 transgenic mice". Journal of Immunology. 165 (4): 1799–806. doi:10.4049/jimmunol.165.4.1799. PMID 10925257.
- Degryse S, de Bock CE, Cox L, Demeyer S, Gielen O, Mentens N, Jacobs K, Geerdens E, Gianfelici V, Hulselmans G, Fiers M, Aerts S, Meijerink JP, Tousseyn T, Cools J (November 2014). "JAK3 mutants transform hematopoietic cells through JAK1 activation, causing T-cell acute lymphoblastic leukemia in a mouse model". Blood. 124 (20): 3092–100. doi:10.1182/blood-2014-04-566687. PMID 25193870.
- Henkels KM, Frondorf K, Gonzalez-Mejia ME, Doseff AL, Gomez-Cambronero J (January 2011). "IL-8-induced neutrophil chemotaxis is mediated by Janus kinase 3 (JAK3)". FEBS Letters. 585 (1): 159–66. doi:10.1016/j.febslet.2010.11.031. PMC 3021320. PMID 21095188.
- Mishra J, Verma RK, Alpini G, Meng F, Kumar N (November 2013). "Role of Janus kinase 3 in mucosal differentiation and predisposition to colitis". The Journal of Biological Chemistry. 288 (44): 31795–806. doi:10.1074/jbc.M113.504126. PMC 3814773. PMID 24045942.
- Mishra J, Kumar N (June 2014). "Adapter protein Shc regulates Janus kinase 3 phosphorylation". The Journal of Biological Chemistry. 289 (23): 15951–6. doi:10.1074/jbc.C113.527523. PMC 4047368. PMID 24795043.
- Mishra J, Verma RK, Alpini G, Meng F, Kumar N (December 2015). "Role of Janus Kinase 3 in Predisposition to Obesity-associated Metabolic Syndrome". The Journal of Biological Chemistry. 290 (49): 29301–12. doi:10.1074/jbc.M115.670331. PMC 4705936. PMID 26451047.
- Mishra, Jayshree; Das, Jugal Kishore; Kumar, Narendra (2017). "Janus kinase 3 regulates adherens junctions and epithelial mesenchymal transition through β-catenin". Journal of Biological Chemistry. 292 (40): 16406–16419. doi:10.1074/jbc.M117.811802. PMC 5633104. PMID 28821617.
- Mishra, Jayshree; Simonsen, Randall; Kumar, Narendra (2019). "Intestinal breast cancer resistance protein (BCRP) requires Janus kinase 3 activity for drug efflux and barrier functions in obesity". Journal of Biological Chemistry. 294. doi:10.1074/jbc.RA119.007758. PMID 31653704.
- Witthuhn BA, Silvennoinen O, Miura O, Lai KS, Cwik C, Liu ET, Ihle JN (July 1994). "Involvement of the Jak-3 Janus kinase in signalling by interleukins 2 and 4 in lymphoid and myeloid cells". Nature. 370 (6485): 153–7. doi:10.1038/370153a0. PMID 8022486.
- Vicente C, Schwab C, Broux M, Geerdens E, Degryse S, Demeyer S, Lahortiga I, Elliott A, Chilton L, La Starza R, Mecucci C, Vandenberghe P, Goulden N, Vora A, Moorman AV, Soulier J, Harrison CJ, Clappier E, Cools J (October 2015). "Targeted sequencing identifies associations between IL7R-JAK mutations and epigenetic modulators in T-cell acute lymphoblastic leukemia". Haematologica. 100 (10): 1301–10. doi:10.3324/haematol.2015.130179. PMC 4591762. PMID 26206799.
- Cox L, Cools J (March 2011). "JAK3 specific kinase inhibitors: when specificity is not enough". Chemistry & Biology. 18 (3): 277–8. doi:10.1016/j.chembiol.2011.03.002. PMID 21439469.
- Suzuki K, Nakajima H, Saito Y, Saito T, Leonard WJ, Iwamoto I (February 2000). "Janus kinase 3 (Jak3) is essential for common cytokine receptor gamma chain (gamma(c))-dependent signaling: comparative analysis of gamma(c), Jak3, and gamma(c) and Jak3 double-deficient mice". International Immunology. 12 (2): 123–32. doi:10.1093/intimm/12.2.123. PMID 10653847.
- O'Shea JJ, Gadina M, Schreiber RD (April 2002). "Cytokine signaling in 2002: new surprises in the Jak/Stat pathway". Cell. 109 (Suppl): S121-31. doi:10.1016/S0092-8674(02)00701-8. PMID 11983158.
- Ferrari-Lacraz S, Zanelli E, Neuberg M, Donskoy E, Kim YS, Zheng XX, Hancock WW, Maslinski W, Li XC, Strom TB, Moll T (November 2004). "Targeting IL-15 receptor-bearing cells with an antagonist mutant IL-15/Fc protein prevents disease development and progression in murine collagen-induced arthritis". Journal of Immunology. 173 (9): 5818–26. doi:10.4049/jimmunol.173.9.5818. PMID 15494535.
- Townsend JM, Fallon GP, Matthews JD, Smith P, Jolin EH, McKenzie NA (October 2000). "IL-9-deficient mice establish fundamental roles for IL-9 in pulmonary mastocytosis and goblet cell hyperplasia but not T cell development" (PDF). Immunity. 13 (4): 573–83. doi:10.1016/S1074-7613(00)00056-X. PMID 11070175.
- West K (May 2009). "CP-690550, a JAK3 inhibitor as an immunosuppressant for the treatment of rheumatoid arthritis, transplant rejection, psoriasis and other immune-mediated disorders". Current Opinion in Investigational Drugs. 10 (5): 491–504. PMID 19431082.
- "Long-Term Effectiveness And Safety Of CP-690,550 For The Treatment Of Rheumatoid Arthritis". ClinicalTrials.gov. 29 February 2012. Retrieved 1 March 2012.
- Tomita K, Saijo K, Yamasaki S, Iida T, Nakatsu F, Arase H, Ohno H, Shirasawa T, Kuriyama T, O'Shea JJ, Saito T (July 2001). "Cytokine-independent Jak3 activation upon T cell receptor (TCR) stimulation through direct association of Jak3 and the TCR complex". The Journal of Biological Chemistry. 276 (27): 25378–85. doi:10.1074/jbc.M011363200. PMID 11349123.
- Ji H, Zhai Q, Zhu J, Yan M, Sun L, Liu X, Zheng Z (April 2000). "A novel protein MAJN binds to Jak3 and inhibits apoptosis induced by IL-2 deprival". Biochemical and Biophysical Research Communications. 270 (1): 267–71. doi:10.1006/bbrc.2000.2413. PMID 10733938.
- Miyazaki T, Kawahara A, Fujii H, Nakagawa Y, Minami Y, Liu ZJ, Oishi I, Silvennoinen O, Witthuhn BA, Ihle JN (November 1994). "Functional activation of Jak1 and Jak3 by selective association with IL-2 receptor subunits". Science. 266 (5187): 1045–7. doi:10.1126/science.7973659. PMID 7973659.
- Russell SM, Johnston JA, Noguchi M, Kawamura M, Bacon CM, Friedmann M, Berg M, McVicar DW, Witthuhn BA, Silvennoinen O (November 1994). "Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: implications for XSCID and XCID". Science. 266 (5187): 1042–5. doi:10.1126/science.7973658. PMID 7973658.
Further reading
- Notarangelo LD, Mella P, Jones A, de Saint Basile G, Savoldi G, Cranston T, Vihinen M, Schumacher RF (October 2001). "Mutations in severe combined immune deficiency (SCID) due to JAK3 deficiency". Human Mutation. 18 (4): 255–63. doi:10.1002/humu.1188. PMID 11668610.
- Russell SM, Tayebi N, Nakajima H, Riedy MC, Roberts JL, Aman MJ, Migone TS, Noguchi M, Markert ML, Buckley RH, O'Shea JJ, Leonard WJ (November 1995). "Mutation of Jak3 in a patient with SCID: essential role of Jak3 in lymphoid development". Science. 270 (5237): 797–800. doi:10.1126/science.270.5237.797. PMID 7481768.
- Johnston JA, Wang LM, Hanson EP, Sun XJ, White MF, Oakes SA, Pierce JH, O'Shea JJ (December 1995). "Interleukins 2, 4, 7, and 15 stimulate tyrosine phosphorylation of insulin receptor substrates 1 and 2 in T cells. Potential role of JAK kinases". The Journal of Biological Chemistry. 270 (48): 28527–30. doi:10.1074/jbc.270.48.28527. PMID 7499365.
- Musso T, Johnston JA, Linnekin D, Varesio L, Rowe TK, O'Shea JJ, McVicar DW (April 1995). "Regulation of JAK3 expression in human monocytes: phosphorylation in response to interleukins 2, 4, and 7". The Journal of Experimental Medicine. 181 (4): 1425–31. doi:10.1084/jem.181.4.1425. PMC 2191962. PMID 7535338.
- Rolling C, Treton D, Beckmann P, Galanaud P, Richard Y (May 1995). "JAK3 associates with the human interleukin 4 receptor and is tyrosine phosphorylated following receptor triggering". Oncogene. 10 (9): 1757–61. PMID 7538655.
- Lai KS, Jin Y, Graham DK, Witthuhn BA, Ihle JN, Liu ET (October 1995). "A kinase-deficient splice variant of the human JAK3 is expressed in hematopoietic and epithelial cancer cells". The Journal of Biological Chemistry. 270 (42): 25028–36. doi:10.1074/jbc.270.42.25028. PMID 7559633.
- Macchi P, Villa A, Giliani S, Sacco MG, Frattini A, Porta F, Ugazio AG, Johnston JA, Candotti F, O'Shea JJ (September 1995). "Mutations of Jak-3 gene in patients with autosomal severe combined immune deficiency (SCID)". Nature. 377 (6544): 65–8. doi:10.1038/377065a0. PMID 7659163.
- Russell SM, Johnston JA, Noguchi M, Kawamura M, Bacon CM, Friedmann M, Berg M, McVicar DW, Witthuhn BA, Silvennoinen O (November 1994). "Interaction of IL-2R beta and gamma c chains with Jak1 and Jak3: implications for XSCID and XCID". Science. 266 (5187): 1042–5. doi:10.1126/science.7973658. PMID 7973658.
- Miyazaki T, Kawahara A, Fujii H, Nakagawa Y, Minami Y, Liu ZJ, Oishi I, Silvennoinen O, Witthuhn BA, Ihle JN (November 1994). "Functional activation of Jak1 and Jak3 by selective association with IL-2 receptor subunits". Science. 266 (5187): 1045–7. doi:10.1126/science.7973659. PMID 7973659.
- Johnston JA, Kawamura M, Kirken RA, Chen YQ, Blake TB, Shibuya K, Ortaldo JR, McVicar DW, O'Shea JJ (July 1994). "Phosphorylation and activation of the Jak-3 Janus kinase in response to interleukin-2". Nature. 370 (6485): 151–3. doi:10.1038/370151a0. PMID 8022485.
- Witthuhn BA, Silvennoinen O, Miura O, Lai KS, Cwik C, Liu ET, Ihle JN (July 1994). "Involvement of the Jak-3 Janus kinase in signalling by interleukins 2 and 4 in lymphoid and myeloid cells". Nature. 370 (6485): 153–7. doi:10.1038/370153a0. PMID 8022486.
- Kawamura M, McVicar DW, Johnston JA, Blake TB, Chen YQ, Lal BK, Lloyd AR, Kelvin DJ, Staples JE, Ortaldo JR (July 1994). "Molecular cloning of L-JAK, a Janus family protein-tyrosine kinase expressed in natural killer cells and activated leukocytes". Proceedings of the National Academy of Sciences of the United States of America. 91 (14): 6374–8. doi:10.1073/pnas.91.14.6374. PMC 44204. PMID 8022790.
- Verbsky JW, Bach EA, Fang YF, Yang L, Randolph DA, Fields LE (June 1996). "Expression of Janus kinase 3 in human endothelial and other non-lymphoid and non-myeloid cells". The Journal of Biological Chemistry. 271 (24): 13976–80. doi:10.1074/jbc.271.24.13976. PMID 8662778.
- Fusaki N, Iwamatsu A, Iwashima M, Fujisawa JI (March 1997). "Interaction between Sam68 and Src family tyrosine kinases, Fyn and Lck, in T cell receptor signaling". The Journal of Biological Chemistry. 272 (10): 6214–9. doi:10.1074/jbc.272.10.6214. PMID 9045636.
- Fujitani Y, Hibi M, Fukada T, Takahashi-Tezuka M, Yoshida H, Yamaguchi T, Sugiyama K, Yamanaka Y, Nakajima K, Hirano T (February 1997). "An alternative pathway for STAT activation that is mediated by the direct interaction between JAK and STAT". Oncogene. 14 (7): 751–61. doi:10.1038/sj.onc.1200907. PMID 9047382.
- Safford MG, Levenstein M, Tsifrina E, Amin S, Hawkins AL, Griffin CA, Civin CI, Small D (May 1997). "JAK3: expression and mapping to chromosome 19p12-13.1". Experimental Hematology. 25 (5): 374–86. PMID 9168059.
- Sharfe N, Dadi HK, O'Shea JJ, Roifman CM (June 1997). "Jak3 activation in human lymphocyte precursor cells". Clinical and Experimental Immunology. 108 (3): 552–6. doi:10.1046/j.1365-2249.1997.4001304.x. PMC 1904698. PMID 9182906.
- Candotti F, Oakes SA, Johnston JA, Giliani S, Schumacher RF, Mella P, Fiorini M, Ugazio AG, Badolato R, Notarangelo LD, Bozzi F, Macchi P, Strina D, Vezzoni P, Blaese RM, O'Shea JJ, Villa A (November 1997). "Structural and functional basis for JAK3-deficient severe combined immunodeficiency". Blood. 90 (10): 3996–4003. PMID 9354668.
External links
- Janus+Kinase+3 at the US National Library of Medicine Medical Subject Headings (MeSH)
- Overview of all the structural information available in the PDB for UniProt: P52333 (Tyrosine-protein kinase JAK3) at the PDBe-KB.
PDB gallery | |
|---|---|
|
