Glycopeptide antibiotic
Glycopeptide antibiotics are a class of drugs of microbial origin that are composed of glycosylated cyclic or polycyclic nonribosomal peptides. Significant glycopeptide antibiotics include the anti-infective antibiotics vancomycin, teicoplanin, telavancin, ramoplanin and decaplanin, corbomycin, complestatin and the antitumor antibiotic bleomycin. Vancomycin is used if infection with methicillin-resistant Staphylococcus aureus (MRSA) is suspected.
| Glycopeptide | |
|---|---|
| Drug class | |
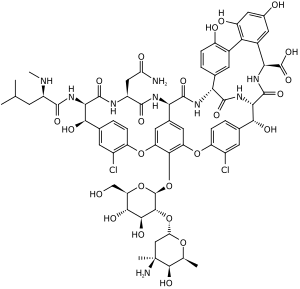 Vancomycin a Glycopeptide | |
| Class identifiers | |
| Use | Bacterial infection |
| ATC code | J01X |
| Biological target | inhibit peptidoglycan synthesis |
| Clinical data | |
| Drugs.com | antibiotics.html Drug Classes |
| In Wikidata | |
Mechanism
Some members of this class of drugs inhibit the synthesis of cell walls in susceptible microbes by inhibiting peptidoglycan synthesis. They bind to the amino acids within the cell wall preventing the addition of new units to the peptidoglycan. In particular, they bind to acyl-D-alanyl-D-alanine in peptidoglycan. Many glycopeptides inhibit the function of glycosyltransferases, which polymerase amino acid/sugar building blocks into peptidoglycan.
Use
Due to their toxicity, use of glycopeptide antibiotics is restricted to patients who are critically ill, who have a demonstrated hypersensitivity to the β-lactams, or who are infected with β-lactam-resistant species. These antibiotics are effective principally against Gram-positive cocci. They exhibit a narrow spectrum of action, and are bactericidal only against the enterococci. Some tissues are not penetrated very well by glycopeptides, and they do not penetrate into the cerebrospinal fluid.
History
Vancomycin was isolated in 1953 and used clinically by 1958, while teicoplanin was discovered in 1978 and became clinically-available in 1984. [1]. Telavancin is a semi-synthetic lipoglycopeptide derivative of vancomycin approved by FDA in 2009.
Teicoplanin has historically been more widely-marketed - and thus more used - in Europe compared to the U.S. It has more fatty acid chains than vancomycin and is considered to be 50 to 100 times more lipophillic. Teicoplanin also has an increased half-life compared to vancomycin, as well as having better tissue penetration. It can be two to four times more active than vancomycin, but it does depend upon the organism. Teicoplanin is more acidic, forming water-soluble salts, so it can be given intramuscularly. Teicoplanin is much better at penetrating into leukocytes and phagocytes than vancomycin.
Since 2002, isolates of vancomycin-resistant Staphylococcus aureus (VRSA) have been found in the USA and other countries.
Glycopeptides have typically been considered the last effective line of defense for cases of MRSA, however several newer classes of antibiotics have proven to have activity against MRSA- including, in 2000, linezolid of the oxazolidinone class, and in 2003 daptomycin of the lipopeptide class.[2]
Research
Several derivatives of vancomycin are currently being developed, including oritavancin and dalbavancin (both lipoglycopeptides). Possessing longer half-lives than vancomycin,[3] these newer candidates may demonstrate improvements over vancomycin due to less frequent dosing and activity against vancomycin-resistant bacteria.
Administration
Vancomycin is usually given intravenously, as an infusion, and can cause tissue necrosis and phlebitis at the injection site if given too rapidly. Pain at site of injection is indeed a common adverse event. One of the side-effects is red man syndrome, an idiosyncratic reaction to bolus caused by histamine release. Some other side-effects of vancomycin are nephrotoxicity including kidney failure and interstitial nephritis, blood disorders including neutropenia, and deafness, which is reversible once therapy has stopped. Over 90% of the dose is excreted in the urine, therefore there is a risk of accumulation in patients with renal impairment, so therapeutic drug monitoring (TDM) is recommended.
Oral preparations of vancomycin are available, however they are not absorbed from the lumen of the gut, so are of no use in treating systemic infections. The oral preparations are formulated for the treatment of infections within the gastrointestinal tract, Clostridium difficile, for example.
References
- Butler MS, Hansford KA, Blaskovich MA, Halai R, Cooper MA (September 2014). "Glycopeptide antibiotics: back to the future". J. Antibiot. 67 (9): 631–44. doi:10.1038/ja.2014.111. PMID 25118105.
- Loffler CA, Macdougall C (December 2007). "Update on prevalence and treatment of methicillin-resistant Staphylococcus aureus infections". Expert Rev Anti Infect Ther. 5 (6): 961–81. doi:10.1586/14787210.5.6.961. PMID 18039081.
- Van Bambeke F. (August 2006). "Glycopeptides and glycodepsipeptides in clinical development: a comparative review of their antibacterial spectrum, pharmacokinetics and clinical efficacy,". Curr Opin Investig Drugs. 7 (8): 740–9. PMID 16955686. http://www.facm.ucl.ac.be/Full-texts-FACM/Vanbambeke-2006-3.pdf