Chlorine
Chlorine is a chemical element with the symbol Cl and atomic number 17. The second-lightest of the halogens, it appears between fluorine and bromine in the periodic table and its properties are mostly intermediate between them. Chlorine is a yellow-green gas at room temperature. It is an extremely reactive element and a strong oxidising agent: among the elements, it has the highest electron affinity and the third-highest electronegativity on the Pauling scale, behind only oxygen and fluorine.
 | |||||||||||||||||||||||
| Chlorine | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pronunciation | /ˈklɔːriːn, -aɪn/ | ||||||||||||||||||||||
| Appearance | pale yellow-green gas | ||||||||||||||||||||||
| Standard atomic weight Ar, std(Cl) | [35.446, 35.457] conventional: 35.45 | ||||||||||||||||||||||
| Chlorine in the periodic table | |||||||||||||||||||||||
| |||||||||||||||||||||||
| Atomic number (Z) | 17 | ||||||||||||||||||||||
| Group | group 17 (halogens) | ||||||||||||||||||||||
| Period | period 3 | ||||||||||||||||||||||
| Block | p-block | ||||||||||||||||||||||
| Element category | Reactive nonmetal | ||||||||||||||||||||||
| Electron configuration | [Ne] 3s2 3p5 | ||||||||||||||||||||||
| Electrons per shell | 2, 8, 7 | ||||||||||||||||||||||
| Physical properties | |||||||||||||||||||||||
| Phase at STP | gas | ||||||||||||||||||||||
| Melting point | (Cl2) 171.6 K (−101.5 °C, −150.7 °F) | ||||||||||||||||||||||
| Boiling point | (Cl2) 239.11 K (−34.04 °C, −29.27 °F) | ||||||||||||||||||||||
| Density (at STP) | 3.2 g/L | ||||||||||||||||||||||
| when liquid (at b.p.) | 1.5625 g/cm3[1] | ||||||||||||||||||||||
| Critical point | 416.9 K, 7.991 MPa | ||||||||||||||||||||||
| Heat of fusion | (Cl2) 6.406 kJ/mol | ||||||||||||||||||||||
| Heat of vaporisation | (Cl2) 20.41 kJ/mol | ||||||||||||||||||||||
| Molar heat capacity | (Cl2) 33.949 J/(mol·K) | ||||||||||||||||||||||
Vapour pressure
| |||||||||||||||||||||||
| Atomic properties | |||||||||||||||||||||||
| Oxidation states | −1, +1, +2, +3, +4, +5, +6, +7 (a strongly acidic oxide) | ||||||||||||||||||||||
| Electronegativity | Pauling scale: 3.16 | ||||||||||||||||||||||
| Ionisation energies |
| ||||||||||||||||||||||
| Covalent radius | 102±4 pm | ||||||||||||||||||||||
| Van der Waals radius | 175 pm | ||||||||||||||||||||||
Color lines in a spectral range | |||||||||||||||||||||||
| Other properties | |||||||||||||||||||||||
| Natural occurrence | primordial | ||||||||||||||||||||||
| Crystal structure | orthorhombic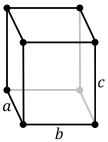 | ||||||||||||||||||||||
| Speed of sound | 206 m/s (gas, at 0 °C) | ||||||||||||||||||||||
| Thermal conductivity | 8.9×10−3 W/(m·K) | ||||||||||||||||||||||
| Electrical resistivity | >10 Ω·m (at 20 °C) | ||||||||||||||||||||||
| Magnetic ordering | diamagnetic[2] | ||||||||||||||||||||||
| Magnetic susceptibility | −40.5·10−6 cm3/mol[3] | ||||||||||||||||||||||
| CAS Number | Cl2: 7782-50-5 | ||||||||||||||||||||||
| History | |||||||||||||||||||||||
| Discovery and first isolation | Carl Wilhelm Scheele (1774) | ||||||||||||||||||||||
| Recognized as an element by | Humphry Davy (1808) | ||||||||||||||||||||||
| Main isotopes of chlorine | |||||||||||||||||||||||
| |||||||||||||||||||||||
The most common compound of chlorine, sodium chloride (common salt), has been known since ancient times. Around 1630, chlorine gas was first synthesised in a chemical reaction, but not recognised as a fundamentally important substance. Carl Wilhelm Scheele wrote a description of chlorine gas in 1774, supposing it to be an oxide of a new element. In 1809, chemists suggested that the gas might be a pure element, and this was confirmed by Sir Humphry Davy in 1810, who named it from Ancient Greek: χλωρός, romanized: khlôros, lit. 'pale green' based on its colour.
Because of its great reactivity, all chlorine in the Earth's crust is in the form of ionic chloride compounds, which includes table salt. It is the second-most abundant halogen (after fluorine) and twenty-first most abundant chemical element in Earth's crust. These crustal deposits are nevertheless dwarfed by the huge reserves of chloride in seawater.
Elemental chlorine is commercially produced from brine by electrolysis, predominantly in the chlor-alkali process. The high oxidising potential of elemental chlorine led to the development of commercial bleaches and disinfectants, and a reagent for many processes in the chemical industry. Chlorine is used in the manufacture of a wide range of consumer products, about two-thirds of them organic chemicals such as polyvinyl chloride (PVC), many intermediates for the production of plastics, and other end products which do not contain the element. As a common disinfectant, elemental chlorine and chlorine-generating compounds are used more directly in swimming pools to keep them sanitary. Elemental chlorine at high concentration is extremely dangerous, and poisonous to most living organisms. As a chemical warfare agent, chlorine was first used in World War I as a poison gas weapon.
In the form of chloride ions, chlorine is necessary to all known species of life. Other types of chlorine compounds are rare in living organisms, and artificially produced chlorinated organics range from inert to toxic. In the upper atmosphere, chlorine-containing organic molecules such as chlorofluorocarbons have been implicated in ozone depletion. Small quantities of elemental chlorine are generated by oxidation of chloride to hypochlorite in neutrophils as part of an immune system response against bacteria.
History

The most common compound of chlorine, sodium chloride, has been known since ancient times; archaeologists have found evidence that rock salt was used as early as 3000 BC and brine as early as 6000 BC.[4] Its importance in food was very well known in classical antiquity and was sometimes used as payment for services for Roman generals and military tribunes. Elemental chlorine was probably first isolated around 1200 with the discovery of aqua regia and its ability to dissolve gold, since chlorine gas is one of the products of this reaction: it was however not recognised as a new substance. Around 1630, chlorine was recognized as a gas by the Flemish chemist and physician Jan Baptist van Helmont.[5][note 1]
The element was first studied in detail in 1774 by Swedish chemist Carl Wilhelm Scheele, and he is credited with the discovery.[6][7] Scheele produced chlorine by reacting MnO2 (as the mineral pyrolusite) with HCl:[5]
- 4 HCl + MnO2 → MnCl2 + 2 H2O + Cl2
Scheele observed several of the properties of chlorine: the bleaching effect on litmus, the deadly effect on insects, the yellow-green color, and the smell similar to aqua regia.[8] He called it "dephlogisticated muriatic acid air" since it is a gas (then called "airs") and it came from hydrochloric acid (then known as "muriatic acid").[7] He failed to establish chlorine as an element.[7]
Common chemical theory at that time held that an acid is a compound that contains oxygen (remnants of this survive in the German and Dutch names of oxygen: sauerstoff or zuurstof, both translating into English as acid substance), so a number of chemists, including Claude Berthollet, suggested that Scheele's dephlogisticated muriatic acid air must be a combination of oxygen and the yet undiscovered element, muriaticum.[9][10]
In 1809, Joseph Louis Gay-Lussac and Louis-Jacques Thénard tried to decompose dephlogisticated muriatic acid air by reacting it with charcoal to release the free element muriaticum (and carbon dioxide).[7] They did not succeed and published a report in which they considered the possibility that dephlogisticated muriatic acid air is an element, but were not convinced.[11]
In 1810, Sir Humphry Davy tried the same experiment again, and concluded that the substance was an element, and not a compound.[7] He announced his results to the Royal Society on 15 November that year.[5] At that time, he named this new element "chlorine", from the Greek word χλωρος (chlōros), meaning green-yellow.[12] The name "halogen", meaning "salt producer", was originally used for chlorine in 1811 by Johann Salomo Christoph Schweigger.[13] This term was later used as a generic term to describe all the elements in the chlorine family (fluorine, bromine, iodine), after a suggestion by Jöns Jakob Berzelius in 1826.[14][15] In 1823, Michael Faraday liquefied chlorine for the first time,[16][17][18] and demonstrated that what was then known as "solid chlorine" had a structure of chlorine hydrate (Cl2·H2O).[5]
Chlorine gas was first used by French chemist Claude Berthollet to bleach textiles in 1785.[19][20] Modern bleaches resulted from further work by Berthollet, who first produced sodium hypochlorite in 1789 in his laboratory in the town of Javel (now part of Paris, France), by passing chlorine gas through a solution of sodium carbonate. The resulting liquid, known as "Eau de Javel" ("Javel water"), was a weak solution of sodium hypochlorite. This process was not very efficient, and alternative production methods were sought. Scottish chemist and industrialist Charles Tennant first produced a solution of calcium hypochlorite ("chlorinated lime"), then solid calcium hypochlorite (bleaching powder).[19] These compounds produced low levels of elemental chlorine and could be more efficiently transported than sodium hypochlorite, which remained as dilute solutions because when purified to eliminate water, it became a dangerously powerful and unstable oxidizer. Near the end of the nineteenth century, E. S. Smith patented a method of sodium hypochlorite production involving electrolysis of brine to produce sodium hydroxide and chlorine gas, which then mixed to form sodium hypochlorite.[21] This is known as the chloralkali process, first introduced on an industrial scale in 1892, and now the source of most elemental chlorine and sodium hydroxide.[22] In 1884 Chemischen Fabrik Griesheim of Germany developed another chloralkali process which entered commercial production in 1888.[23]
Elemental chlorine solutions dissolved in chemically basic water (sodium and calcium hypochlorite) were first used as anti-putrefaction agents and disinfectants in the 1820s, in France, long before the establishment of the germ theory of disease. This practice was pioneered by Antoine-Germain Labarraque, who adapted Berthollet's "Javel water" bleach and other chlorine preparations (for a more complete history, see below).[24] Elemental chlorine has since served a continuous function in topical antisepsis (wound irrigation solutions and the like) and public sanitation, particularly in swimming and drinking water.[8]
Chlorine gas was first used as a weapon on April 22, 1915, at Ypres by the German Army.[25][26] The effect on the allies was devastating because the existing gas masks were difficult to deploy and had not been broadly distributed.[27][28]
Properties


Chlorine is the second halogen, being a nonmetal in group 17 of the periodic table. Its properties are thus similar to fluorine, bromine, and iodine, and are largely intermediate between those of the first two. Chlorine has the electron configuration [Ne]3s23p5, with the seven electrons in the third and outermost shell acting as its valence electrons. Like all halogens, it is thus one electron short of a full octet, and is hence a strong oxidising agent, reacting with many elements in order to complete its outer shell.[29] Corresponding to periodic trends, it is intermediate in electronegativity between fluorine and bromine (F: 3.98, Cl: 3.16, Br: 2.96, I: 2.66), and is less reactive than fluorine and more reactive than bromine. It is also a weaker oxidising agent than fluorine, but a stronger one than bromine. Conversely, the chloride ion is a weaker reducing agent than bromide, but a stronger one than fluoride.[29] It is intermediate in atomic radius between fluorine and bromine, and this leads to many of its atomic properties similarly continuing the trend from iodine to bromine upward, such as first ionisation energy, electron affinity, enthalpy of dissociation of the X2 molecule (X = Cl, Br, I), ionic radius, and X–X bond length. (Fluorine is anomalous due to its small size.)[29]
All four stable halogens experience intermolecular van der Waals forces of attraction, and their strength increases together with the number of electrons among all homonuclear diatomic halogen molecules. Thus, the melting and boiling points of chlorine are intermediate between those of fluorine and bromine: chlorine melts at −101.0 °C and boils at −34.0 °C. As a result of the increasing molecular weight of the halogens down the group, the density and heats of fusion and vaporisation of chlorine are again intermediate between those of bromine and fluorine, although all their heats of vaporisation are fairly low (leading to high volatility) thanks to their diatomic molecular structure.[29] The halogens darken in colour as the group is descended: thus, while fluorine is a pale yellow gas, chlorine is distinctly yellow-green. This trend occurs because the wavelengths of visible light absorbed by the halogens increase down the group.[29] Specifically, the colour of a halogen, such as chlorine, results from the electron transition between the highest occupied antibonding πg molecular orbital and the lowest vacant antibonding σu molecular orbital.[30] The colour fades at low temperatures, so that solid chlorine at −195 °C is almost colourless.[29]
Like solid bromine and iodine, solid chlorine crystallises in the orthorhombic crystal system, in a layered lattice of Cl2 molecules. The Cl–Cl distance is 198 pm (close to the gaseous Cl–Cl distance of 199 pm) and the Cl···Cl distance between molecules is 332 pm within a layer and 382 pm between layers (compare the van der Waals radius of chlorine, 180 pm). This structure means that chlorine is a very poor conductor of electricity, and indeed its conductivity is so low as to be practically unmeasurable.[29]
Isotopes
Chlorine has two stable isotopes, 35Cl and 37Cl. These are its only two natural isotopes occurring in quantity, with 35Cl making up 76% of natural chlorine and 37Cl making up the remaining 24%. Both are synthesised in stars in the oxygen-burning and silicon-burning processes.[31] Both have nuclear spin 3/2+ and thus may be used for nuclear magnetic resonance, although the spin magnitude being greater than 1/2 results in non-spherical nuclear charge distribution and thus resonance broadening as a result of a nonzero nuclear quadrupole moment and resultant quadrupolar relaxation. The other chlorine isotopes are all radioactive, with half-lives too short to occur in nature primordially. Of these, the most commonly used in the laboratory are 36Cl (t1/2 = 3.0×105 y) and 38Cl (t1/2 = 37.2 min), which may be produced from the neutron activation of natural chlorine.[29]
The most stable chlorine radioisotope is 36Cl. The primary decay mode of isotopes lighter than 35Cl is electron capture to isotopes of sulfur; that of isotopes heavier than 37Cl is beta decay to isotopes of argon; and 36Cl may decay by either mode to stable 36S or 36Ar.[32] 36Cl occurs in trace quantities in nature as a cosmogenic nuclide in a ratio of about (7–10) × 10−13 to 1 with stable chlorine isotopes: it is produced in the atmosphere by spallation of 36Ar by interactions with cosmic ray protons. In the top meter of the lithosphere, 36Cl is generated primarily by thermal neutron activation of 35Cl and spallation of 39K and 40Ca. In the subsurface environment, muon capture by 40Ca becomes more important as a way to generate 36Cl.[33][34]
Chemistry and compounds
| X | XX | HX | BX3 | AlX3 | CX4 |
|---|---|---|---|---|---|
| F | 159 | 574 | 645 | 582 | 456 |
| Cl | 243 | 428 | 444 | 427 | 327 |
| Br | 193 | 363 | 368 | 360 | 272 |
| I | 151 | 294 | 272 | 285 | 239 |
Chlorine is intermediate in reactivity between fluorine and bromine, and is one of the most reactive elements. Chlorine is a weaker oxidising agent than fluorine but a stronger one than bromine or iodine. This can be seen from the standard electrode potentials of the X2/X− couples (F, +2.866 V; Cl, +1.395 V; Br, +1.087 V; I, +0.615 V; At, approximately +0.3 V). However, this trend is not shown in the bond energies because fluorine is singular due to its small size, low polarisability, and lack of low-lying d-orbitals available for bonding (which chlorine has). As another difference, chlorine has a significant chemistry in positive oxidation states while fluorine does not. Chlorination often leads to higher oxidation states than bromination or iodination but lower oxidation states than fluorination. Chlorine tends to react with compounds including M–M, M–H, or M–C bonds to form M–Cl bonds.[30]
Given that E°(1/2O2/H2O) = +1.229 V, which is less than +1.395 V, it would be expected that chlorine should be able to oxidise water to oxygen and hydrochloric acid. However, the kinetics of this reaction are unfavorable, and there is also a bubble overpotential effect to consider, so that electrolysis of aqueous chloride solutions evolves chlorine gas and not oxygen gas, a fact that is very useful for the industrial production of chlorine.[35]
Hydrogen chloride
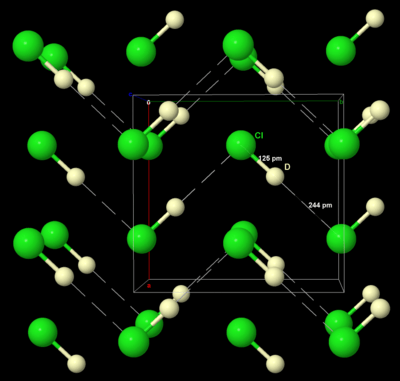
The simplest chlorine compound is hydrogen chloride, HCl, a major chemical in industry as well as in the laboratory, both as a gas and dissolved in water as hydrochloric acid. It is often produced by burning hydrogen gas in chlorine gas, or as a byproduct of chlorinating hydrocarbons. Another approach is to treat sodium chloride with concentrated sulfuric acid to produce hydrochloric acid, also known as the "salt-cake" process:[36]
- NaCl + H2SO4 NaHSO4 + HCl
- NaCl + NaHSO4 Na2SO4 + HCl
In the laboratory, hydrogen chloride gas may be made by drying the acid with concentrated sulfuric acid. Deuterium chloride, DCl, may be produced by reacting benzoyl chloride with heavy water (D2O).[36]
At room temperature, hydrogen chloride is a colourless gas, like all the hydrogen halides apart from hydrogen fluoride, since hydrogen cannot form strong hydrogen bonds to the larger electronegative chlorine atom; however, weak hydrogen bonding is present in solid crystalline hydrogen chloride at low temperatures, similar to the hydrogen fluoride structure, before disorder begins to prevail as the temperature is raised.[36] Hydrochloric acid is a strong acid (pKa = −7) because the hydrogen bonds to chlorine are too weak to inhibit dissociation. The HCl/H2O system has many hydrates HCl·nH2O for n = 1, 2, 3, 4, and 6. Beyond a 1:1 mixture of HCl and H2O, the system separates completely into two separate liquid phases. Hydrochloric acid forms an azeotrope with boiling point 108.58 °C at 20.22 g HCl per 100 g solution; thus hydrochloric acid cannot be concentrated beyond this point by distillation.[37]
Unlike hydrogen fluoride, anhydrous liquid hydrogen chloride is difficult to work with as a solvent, because its boiling point is low, it has a small liquid range, its dielectric constant is low and it does not dissociate appreciably into H2Cl+ and HCl−
2 ions – the latter, in any case, are much less stable than the bifluoride ions (HF−
2) due to the very weak hydrogen bonding between hydrogen and chlorine, though its salts with very large and weakly polarising cations such as Cs+ and NR+
4 (R = Me, Et, Bun) may still be isolated. Anhydrous hydrogen chloride is a poor solvent, only able to dissolve small molecular compounds such as nitrosyl chloride and phenol, or salts with very low lattice energies such as tetraalkylammonium halides. It readily protonates electrophiles containing lone-pairs or π bonds. Solvolysis, ligand replacement reactions, and oxidations are well-characterised in hydrogen chloride solution:[38]
- Ph3SnCl + HCl ⟶ Ph2SnCl2 + PhH (solvolysis)
- Ph3COH + 3 HCl ⟶ Ph
3C+
HCl−
2 + H3O+Cl− (solvolysis) - Me
4N+
HCl−
2 + BCl3 ⟶ Me
4N+
BCl−
4 + HCl (ligand replacement) - PCl3 + Cl2 + HCl ⟶ PCl+
4HCl−
2 (oxidation)
Other binary chlorides
-chloride-hexahydrate-sample.jpg)
Nearly all elements in the periodic table form binary chlorides. The exceptions are decidedly in the minority and stem in each case from one of three causes: extreme inertness and reluctance to participate in chemical reactions (the noble gases, with the exception of xenon in the highly unstable XeCl2 and XeCl4); extreme nuclear instability hampering chemical investigation before decay and transmutation (many of the heaviest elements beyond bismuth); and having an electronegativity higher than chlorine's (oxygen and fluorine) so that the resultant binary compounds are formally not chlorides but rather oxides or fluorides of chlorine.[39]
Chlorination of metals with Cl2 usually leads to a higher oxidation state than bromination with Br2 when multiple oxidation states are available, such as in MoCl5 and MoBr3. Chlorides can be made by reaction of an element or its oxide, hydroxide, or carbonate with hydrochloric acid, and then dehydrated by mildly high temperatures combined with either low pressure or anhydrous hydrogen chloride gas. These methods work best when the chloride product is stable to hydrolysis; otherwise, the possibilities include high-temperature oxidative chlorination of the element with chlorine or hydrogen chloride, high-temperature chlorination of a metal oxide or other halide by chlorine, a volatile metal chloride, carbon tetrachloride, or an organic chloride. For instance, zirconium dioxide reacts with chlorine at standard conditions to produce zirconium tetrachloride, and uranium trioxide reacts with hexachloropropene when heated under reflux to give uranium tetrachloride. The second example also involves a reduction in oxidation state, which can also be achieved by reducing a higher chloride using hydrogen or a metal as a reducing agent. This may also be achieved by thermal decomposition or disproportionation as follows:[39]
- EuCl3 + 1/2 H2 ⟶ EuCl2 + HCl
- ReCl5 ReCl3 + Cl2
- AuCl3 AuCl + Cl2
Most of the chlorides of the pre-transition metals (groups 1, 2, and 3, along with the lanthanides and actinides in the +2 and +3 oxidation states) are mostly ionic, while nonmetals tend to form covalent molecular chlorides, as do metals in high oxidation states from +3 and above. Silver chloride is very insoluble in water and is thus often used as a qualitative test for chlorine.[39]
Polychlorine compounds
Although dichlorine is a strong oxidising agent with a high first ionisation energy, it may be oxidised under extreme conditions to form the Cl+
2 cation. This is very unstable and has only been characterised by its electronic band spectrum when produced in a low-pressure discharge tube. The yellow Cl+
3 cation is more stable and may be produced as follows:[40]
- Cl2 + ClF + AsF5 Cl+
3AsF−
6
This reaction is conducted in the oxidising solvent arsenic pentafluoride. The trichloride anion, Cl−
3, has also been characterised; it is analogous to triiodide.[41]
Chlorine fluorides
The three fluorides of chlorine form a subset of the interhalogen compounds, all of which are diamagnetic.[41] Some cationic and anionic derivatives are known, such as ClF−
2, ClF−
4, ClF+
2, and Cl2F+.[42] Some pseudohalides of chlorine are also known, such as cyanogen chloride (ClCN, linear), chlorine cyanate (ClNCO), chlorine thiocyanate (ClSCN, unlike its oxygen counterpart), and chlorine azide (ClN3).[41]
Chlorine monofluoride (ClF) is extremely thermally stable, and is sold commercially in 500-gram steel lecture bottles. It is a colourless gas that melts at −155.6 °C and boils at −100.1 °C. It may be produced by the direction of its elements at 225 °C, though it must then be separated and purified from chlorine trifluoride and its reactants. Its properties are mostly intermediate between those of chlorine and fluorine. It will react with many metals and nonmetals from room temperature and above, fluorinating them and liberating chlorine. It will also act as a chlorofluorinating agent, adding chlorine and fluorine across a multiple bond or by oxidation: for example, it will attack carbon monoxide to form carbonyl chlorofluoride, COFCl. It will react analogously with hexafluoroacetone, (CF3)2CO, with a potassium fluoride catalyst to produce heptafluoroisopropyl hypochlorite, (CF3)2CFOCl; with nitriles RCN to produce RCF2NCl2; and with the sulfur oxides SO2 and SO3 to produce ClOSO2F and ClSO2F respectively. It will also react exothermically and violently with compounds containing –OH and –NH groups, such as water:[41]
- H2O + 2 ClF ⟶ 2 HF + Cl2O
Chlorine trifluoride (ClF3) is a volatile colourless molecular liquid which melts at −76.3 °C and boils at 11.8 °C. It may be formed by directly fluorinating gaseous chlorine or chlorine monofluoride at 200–300 °C. It is one of the most reactive known chemical compounds, reacting with many substances which in ordinary circumstances would be considered chemically inert, such as asbestos, concrete, and sand. It explodes on contact with water and most organic substances. The list of elements it sets on fire is diverse, containing hydrogen, potassium, phosphorus, arsenic, antimony, sulfur, selenium, tellurium, bromine, iodine, and powdered molybdenum, tungsten, rhodium, iridium, and iron. An impermeable fluoride layer is formed by sodium, magnesium, aluminium, zinc, tin, and silver, which may be removed by heating. When heated, even such noble metals as palladium, platinum, and gold are attacked and even the noble gases xenon and radon do not escape fluorination. Nickel containers are usually used due to that metal's great resistance to attack by chlorine trifluoride, stemming from the formation of an unreactive nickel fluoride layer. Its reaction with hydrazine to form hydrogen fluoride, nitrogen, and chlorine gases was used in experimental rocket motors, but has problems largely stemming from its extreme hypergolicity resulting in ignition without any measurable delay. Today, it is mostly used in nuclear fuel processing, to oxidise uranium to uranium hexafluoride for its enriching and to separate it from plutonium. It can act as a fluoride ion donor or acceptor (Lewis base or acid), although it does not dissociate appreciably into ClF+
2 and ClF−
4 ions.[43]
Chlorine pentafluoride (ClF5) is made on a large scale by direct fluorination of chlorine with excess fluorine gas at 350 °C and 250 atm, and on a small scale by reacting metal chlorides with fluorine gas at 100–300 °C. It melts at −103 °C and boils at −13.1 °C. It is a very strong fluorinating agent, although it is still not as effective as chlorine trifluoride. Only a few specific stoichiometric reactions have been characterised. Arsenic pentafluoride and antimony pentafluoride form ionic adducts of the form [ClF4]+[MF6]− (M = As, Sb) and water reacts vigorously as follows:[44]
- 2 H2O + ClF5 ⟶ 4 HF + FClO2
The product, chloryl fluoride, is one of the five known chlorine oxide fluorides. These range from the thermally unstable FClO to the chemically unreactive perchloryl fluoride (FClO3), the other three being FClO2, F3ClO, and F3ClO2. All five behave similarly to the chlorine fluorides, both structurally and chemically, and may act as Lewis acids or bases by gaining or losing fluoride ions respectively or as very strong oxidising and fluorinating agents.[45]
Chlorine oxides

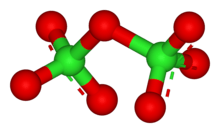
The chlorine oxides are well-studied in spite of their instability (all of them are endothermic compounds). They are important because they are produced when chlorofluorocarbons undergo photolysis in the upper atmosphere and cause the destruction of the ozone layer. None of them can be made from directly reacting the elements.[46]
Dichlorine monoxide (Cl2O) is a brownish-yellow gas (red-brown when solid or liquid) which may be obtained by reacting chlorine gas with yellow mercury(II) oxide. It is very soluble in water, in which it is in equilibrium with hypochlorous acid (HOCl), of which it is the anhydride. It is thus an effective bleach and is mostly used to make hypochlorites. It explodes on heating or sparking or in the presence of ammonia gas.[46]
Chlorine dioxide (ClO2) was the first chlorine oxide to be discovered in 1811 by Humphry Davy. It is a yellow paramagnetic gas (deep-red as a solid or liquid), as expected from its having an odd number of electrons: it is stable towards dimerisation due to the delocalisation of the unpaired electron. It explodes above −40 °C as a liquid and under pressure as a gas and therefore must be made at low concentrations for wood-pulp bleaching and water treatment. It is usually prepared by reducing a chlorate as follows:[46]
- ClO−
3 + Cl− + 2 H+ ⟶ ClO2 + 1/2 Cl2 + H2O
Its production is thus intimately linked to the redox reactions of the chlorine oxoacids. It is a strong oxidising agent, reacting with sulfur, phosphorus, phosphorus halides, and potassium borohydride. It dissolves exothermically in water to form dark-green solutions that very slowly decompose in the dark. Crystalline clathrate hydrates ClO2·nH2O (n ≈ 6–10) separate out at low temperatures. However, in the presence of light, these solutions rapidly photodecompose to form a mixture of chloric and hydrochloric acids. Photolysis of individual ClO2 molecules result in the radicals ClO and ClOO, while at room temperature mostly chlorine, oxygen, and some ClO3 and Cl2O6 are produced. Cl2O3 is also produced when photolysing the solid at −78 °C: it is a dark brown solid that explodes below 0 °C. The ClO radical leads to the depletion of atmospheric ozone and is thus environmentally important as follows:[46]
- Cl• + O3 ⟶ ClO• + O2
- ClO• + O• ⟶ Cl• + O2
Chlorine perchlorate (ClOClO3) is a pale yellow liquid that is less stable than ClO2 and decomposes at room temperature to form chlorine, oxygen, and dichlorine hexoxide (Cl2O6).[46] Chlorine perchlorate may also be considered a chlorine derivative of perchloric acid (HOClO3), similar to the thermally unstable chlorine derivatives of other oxoacids: examples include chlorine nitrate (ClONO2, vigorously reactive and explosive), and chlorine fluorosulfate (ClOSO2F, more stable but still moisture-sensitive and highly reactive).[47] Dichlorine hexoxide is a dark-red liquid that freezes to form a solid which turns yellow at −180 °C: it is usually made by reaction of chlorine dioxide with oxygen. Despite attempts to rationalise it as the dimer of ClO3, it reacts more as though it were chloryl perchlorate, [ClO2]+[ClO4]−, which has been confirmed to be the correct structure of the solid. It hydrolyses in water to give a mixture of chloric and perchloric acids: the analogous reaction with anhydrous hydrogen fluoride does not proceed to completion.[46]
Dichlorine heptoxide (Cl2O7) is the anhydride of perchloric acid (HClO4) and can readily be obtained from it by dehydrating it with phosphoric acid at −10 °C and then distilling the product at −35 °C and 1 mmHg. It is a shock-sensitive, colourless oily liquid. It is the least reactive of the chlorine oxides, being the only one to not set organic materials on fire at room temperature. It may be dissolved in water to regenerate perchloric acid or in aqueous alkalis to regenerate perchlorates. However, it thermally decomposes explosively by breaking one of the central Cl–O bonds, producing the radicals ClO3 and ClO4 which immediately decompose to the elements through intermediate oxides.[46]
Chlorine oxoacids and oxyanions
| E°(couple) | a(H+) = 1 (acid) | E°(couple) | a(OH−) = 1 (base) |
|---|---|---|---|
| Cl2/Cl− | +1.358 | Cl2/Cl− | +1.358 |
| HOCl/Cl− | +1.484 | ClO−/Cl− | +0.890 |
| ClO− 3/Cl− | +1.459 | ||
| HOCl/Cl2 | +1.630 | ClO−/Cl2 | +0.421 |
| HClO2/Cl2 | +1.659 | ||
| ClO− 3/Cl2 | +1.468 | ||
| ClO− 4/Cl2 | +1.277 | ||
| HClO2/HOCl | +1.701 | ClO− 2/ClO− | +0.681 |
| ClO− 3/ClO− | +0.488 | ||
| ClO− 3/HClO2 | +1.181 | ClO− 3/ClO− 2 | +0.295 |
| ClO− 4/ClO− 3 | +1.201 | ClO− 4/ClO− 3 | +0.374 |
Chlorine forms four oxoacids: hypochlorous acid (HOCl), chlorous acid (HOClO), chloric acid (HOClO2), and perchloric acid (HOClO3). As can be seen from the redox potentials given in the adjacent table, chlorine is much more stable towards disproportionation in acidic solutions than in alkaline solutions:[35]
Cl2 + H2O ⇌ HOCl + H+ + Cl− Kac = 4.2 × 10−4 mol2 l−2 Cl2 + 2 OH− ⇌ OCl− + H2O + Cl− Kalk = 7.5 × 1015 mol−1 l
The hypochlorite ions also disproportionate further to produce chloride and chlorate (3 ClO− ⇌ 2 Cl− + ClO−
3) but this reaction is quite slow at temperatures below 70 °C in spite of the very favourable equilibrium constant of 1027. The chlorate ions may themselves disproportionate to form chloride and perchlorate (4 ClO−
3 ⇌ Cl− + 3 ClO−
4) but this is still very slow even at 100 °C despite the very favourable equilibrium constant of 1020. The rates of reaction for the chlorine oxyanions increases as the oxidation state of chlorine decreases. The strengths of the chlorine oxyacids increase very quickly as the oxidation state of chlorine increases due to the increasing delocalisation of charge over more and more oxygen atoms in their conjugate bases.[35]
Most of the chlorine oxoacids may be produced by exploiting these disproportionation reactions. Hypochlorous acid (HOCl) is highly reactive and quite unstable; its salts are mostly used for their bleaching and sterilising abilities. They are very strong oxidising agents, transferring an oxygen atom to most inorganic species. Chlorous acid (HOClO) is even more unstable and cannot be isolated or concentrated without decomposition: it is known from the decomposition of aqueous chlorine dioxide. However, sodium chlorite is a stable salt and is useful for bleaching and stripping textiles, as an oxidising agent, and as a source of chlorine dioxide. Chloric acid (HOClO2) is a strong acid that is quite stable in cold water up to 30% concentration, but on warming gives chlorine and chlorine dioxide. Evaporation under reduced pressure allows it to be concentrated further to about 40%, but then it decomposes to perchloric acid, chlorine, oxygen, water, and chlorine dioxide. Its most important salt is sodium chlorate, mostly used to make chlorine dioxide to bleach paper pulp. The decomposition of chlorate to chloride and oxygen is a common way to produce oxygen in the laboratory on a small scale. Chloride and chlorate may comproportionate to form chlorine as follows:[48]
- ClO−
3 + 5 Cl− + 6 H+ ⟶ 3 Cl2 + 3 H2O
Perchlorates and perchloric acid (HOClO3) are the most stable oxo-compounds of chlorine, in keeping with the fact that chlorine compounds are most stable when the chlorine atom is in its lowest (−1) or highest (+7) possible oxidation states. Perchloric acid and aqueous perchlorates are vigorous and sometimes violent oxidising agents when heated, in stark contrast to their mostly inactive nature at room temperature due to the high activation energies for these reactions for kinetic reasons. Perchlorates are made by electrolytically oxidising sodium chlorate, and perchloric acid is made by reacting anhydrous sodium perchlorate or barium perchlorate with concentrated hydrochloric acid, filtering away the chloride precipitated and distilling the filtrate to concentrate it. Anhydrous perchloric acid is a colourless mobile liquid that is sensitive to shock that explodes on contact with most organic compounds, sets hydrogen iodide and thionyl chloride on fire and even oxidises silver and gold. Although it is a weak ligand, weaker than water, a few compounds involving coordinated ClO−
4 are known.[48]
Organochlorine compounds
Like the other carbon–halogen bonds, the C–Cl bond is a common functional group that forms part of core organic chemistry. Formally, compounds with this functional group may be considered organic derivatives of the chloride anion. Due to the difference of electronegativity between chlorine (3.16) and carbon (2.55), the carbon in a C–Cl bond is electron-deficient and thus electrophilic. Chlorination modifies the physical properties of hydrocarbons in several ways: chlorocarbons are typically denser than water due to the higher atomic weight of chlorine versus hydrogen, and aliphatic organochlorides are alkylating agents because chloride is a leaving group.[49]
Alkanes and aryl alkanes may be chlorinated under free-radical conditions, with UV light. However, the extent of chlorination is difficult to control: the reaction is not regioselective and often results in a mixture of various isomers with different degrees of chlorination, though this may be permissible if the products are easily separated. Aryl chlorides may be prepared by the Friedel-Crafts halogenation, using chlorine and a Lewis acid catalyst.[49] The haloform reaction, using chlorine and sodium hydroxide, is also able to generate alkyl halides from methyl ketones, and related compounds. Chlorine adds to the multiple bonds on alkenes and alkynes as well, giving di- or tetra-chloro compounds. However, due to the expense and reactivity of chlorine, organochlorine compounds are more commonly produced by using hydrogen chloride, or with chlorinating agents such as phosphorus pentachloride (PCl5) or thionyl chloride (SOCl2). The last is very convenient in the laboratory because all side products are gaseous and do not have to be distilled out.[49]
Many organochlorine compounds have been isolated from natural sources ranging from bacteria to humans.[50][51] Chlorinated organic compounds are found in nearly every class of biomolecules including alkaloids, terpenes, amino acids, flavonoids, steroids, and fatty acids.[50][52] Organochlorides, including dioxins, are produced in the high temperature environment of forest fires, and dioxins have been found in the preserved ashes of lightning-ignited fires that predate synthetic dioxins.[53] In addition, a variety of simple chlorinated hydrocarbons including dichloromethane, chloroform, and carbon tetrachloride have been isolated from marine algae.[54] A majority of the chloromethane in the environment is produced naturally by biological decomposition, forest fires, and volcanoes.[55]
Some types of organochlorides, though not all, have significant toxicity to plants or animals, including humans. Dioxins, produced when organic matter is burned in the presence of chlorine, and some insecticides, such as DDT, are persistent organic pollutants which pose dangers when they are released into the environment. For example, DDT, which was widely used to control insects in the mid 20th century, also accumulates in food chains, and causes reproductive problems (e.g., eggshell thinning) in certain bird species.[56] Due to the ready homolytic fission of the C–Cl bond to create chlorine radicals in the upper atmosphere, chlorofluorocarbons have been phased out due to the harm they do to the ozone layer.[46]
Occurrence and production

Chlorine is too reactive to occur as the free element in nature but is very abundant in the form of its chloride salts. It is the twenty-first most abundant element in Earth's crust and makes up 126 parts per million of it, through the large deposits of chloride minerals, especially sodium chloride, that have been evaporated from water bodies. All of these pale in comparison to the reserves of chloride ions in seawater: smaller amounts at higher concentrations occur in some inland seas and underground brine wells, such as the Great Salt Lake in Utah and the Dead Sea in Israel.[57]
Small batches of chlorine gas are prepared in the laboratory by combining hydrochloric acid and manganese dioxide, but the need rarely arises due to its ready availability. In industry, elemental chlorine is usually produced by the electrolysis of sodium chloride dissolved in water. This method, the chloralkali process industrialized in 1892, now provides most industrial chlorine gas.[22] Along with chlorine, the method yields hydrogen gas and sodium hydroxide, which is the most valuable product. The process proceeds according to the following chemical equation:[58]
- 2 NaCl + 2 H2O → Cl2 + H2 + 2 NaOH
The electrolysis of chloride solutions all proceed according to the following equations:
- Cathode: 2 H2O + 2 e− → H2 + 2 OH−
- Anode: 2 Cl− → Cl2 + 2 e−
In diaphragm cell electrolysis, an asbestos (or polymer-fiber) diaphragm separates a cathode and an anode, preventing the chlorine forming at the anode from re-mixing with the sodium hydroxide and the hydrogen formed at the cathode.[59] The salt solution (brine) is continuously fed to the anode compartment and flows through the diaphragm to the cathode compartment, where the caustic alkali is produced and the brine is partially depleted. Diaphragm methods produce dilute and slightly impure alkali, but they are not burdened with the problem of mercury disposal and they are more energy efficient.[22]
Membrane cell electrolysis employs permeable membrane as an ion exchanger. Saturated sodium (or potassium) chloride solution is passed through the anode compartment, leaving at a lower concentration. This method also produces very pure sodium (or potassium) hydroxide but has the disadvantage of requiring very pure brine at high concentrations.[60]
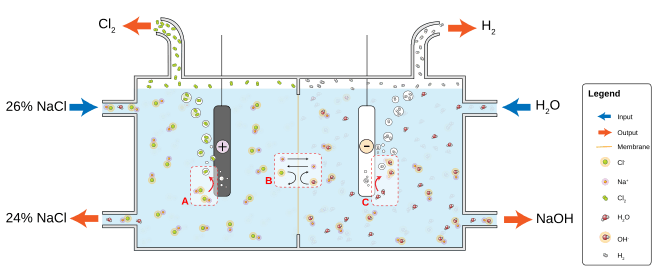
In the Deacon process, hydrogen chloride recovered from the production of organochlorine compounds is recovered as chlorine. The process relies on oxidation using oxygen:
- 4 HCl + O2 → 2 Cl2 + 2 H2O
The reaction requires a catalyst. As introduced by Deacon, early catalysts were based on copper. Commercial processes, such as the Mitsui MT-Chlorine Process, have switched to chromium and ruthenium-based catalysts.[61] The chlorine produced is available in cylinders from sizes ranging from 450 g to 70 kg, as well as drums (865 kg), tank wagons (15 tonnes on roads; 27–90 tonnes by rail), and barges (600–1200 tonnes).[62]
Applications
Sodium chloride is the most common chlorine compound, and is the main source of chlorine for the enormous demand associated with today's chemicals industry. About 15000 chlorine-containing compounds are commercially traded, including such diverse compounds as chlorinated methane, ethanes, vinyl chloride, polyvinyl chloride (PVC), aluminium trichloride for catalysis, the chlorides of magnesium, titanium, zirconium, and hafnium which are the precursors for producing the pure form of those elements.[8]
Quantitatively, of all elemental chlorine produced, about 63% is used in the manufacture of organic compounds, and 18% in the manufacture of inorganic chlorine compounds.[63] About 15,000 chlorine compounds are used commercially.[64] The remaining 19% of chlorine produced is used for bleaches and disinfection products.[62] The most significant of organic compounds in terms of production volume are 1,2-dichloroethane and vinyl chloride, intermediates in the production of PVC. Other particularly important organochlorines are methyl chloride, methylene chloride, chloroform, vinylidene chloride, trichloroethylene, perchloroethylene, allyl chloride, epichlorohydrin, chlorobenzene, dichlorobenzenes, and trichlorobenzenes. The major inorganic compounds include HCl, Cl2O, HOCl, NaClO3, chlorinated isocyanurates, AlCl3, SiCl4, SnCl4, PCl3, PCl5, POCl3, AsCl3, SbCl3, SbCl5, BiCl3, S2Cl2, SCl2, SOCI2, ClF3, ICl, ICl3, TiCl3, TiCl4, MoCl5, FeCl3, ZnCl2, and so on.[62]
Sanitation, disinfection, and antisepsis
Combating putrefaction

In France (as elsewhere), animal intestines were processed to make musical instrument strings, Goldbeater's skin and other products. This was done in "gut factories" (boyauderies), and it was an odiferous and unhealthy process. In or about 1820, the Société d'encouragement pour l'industrie nationale offered a prize for the discovery of a method, chemical or mechanical, for separating the peritoneal membrane of animal intestines without putrefaction.[65][66] The prize was won by Antoine-Germain Labarraque, a 44-year-old French chemist and pharmacist who had discovered that Berthollet's chlorinated bleaching solutions ("Eau de Javel") not only destroyed the smell of putrefaction of animal tissue decomposition, but also actually retarded the decomposition.[66][67]
Labarraque's research resulted in the use of chlorides and hypochlorites of lime (calcium hypochlorite) and of sodium (sodium hypochlorite) in the boyauderies. The same chemicals were found to be useful in the routine disinfection and deodorization of latrines, sewers, markets, abattoirs, anatomical theatres, and morgues.[68] They were successful in hospitals, lazarets, prisons, infirmaries (both on land and at sea), magnaneries, stables, cattle-sheds, etc.; and they were beneficial during exhumations,[69] embalming, outbreaks of epidemic disease, fever, and blackleg in cattle.[65]
Disinfection
Labarraque's chlorinated lime and soda solutions have been advocated since 1828 to prevent infection (called "contagious infection", presumed to be transmitted by "miasmas"), and to treat putrefaction of existing wounds, including septic wounds.[70] In his 1828 work, Labarraque recommended that doctors breathe chlorine, wash their hands in chlorinated lime, and even sprinkle chlorinated lime about the patients' beds in cases of "contagious infection". In 1828, the contagion of infections was well known, even though the agency of the microbe was not discovered until more than half a century later.
During the Paris cholera outbreak of 1832, large quantities of so-called chloride of lime were used to disinfect the capital. This was not simply modern calcium chloride, but chlorine gas dissolved in lime-water (dilute calcium hydroxide) to form calcium hypochlorite (chlorinated lime). Labarraque's discovery helped to remove the terrible stench of decay from hospitals and dissecting rooms, and by doing so, effectively deodorised the Latin Quarter of Paris.[71] These "putrid miasmas" were thought by many to cause the spread of "contagion" and "infection" – both words used before the germ theory of infection. Chloride of lime was used for destroying odors and "putrid matter". One source claims chloride of lime was used by Dr. John Snow to disinfect water from the cholera-contaminated well that was feeding the Broad Street pump in 1854 London,[72] though three other reputable sources that describe that famous cholera epidemic do not mention the incident.[73][74][75] One reference makes it clear that chloride of lime was used to disinfect the offal and filth in the streets surrounding the Broad Street pump—a common practice in mid-nineteenth century England.[73]:296
Semmelweis and experiments with antisepsis

Perhaps the most famous application of Labarraque's chlorine and chemical base solutions was in 1847, when Ignaz Semmelweis used chlorine-water (chlorine dissolved in pure water, which was cheaper than chlorinated lime solutions) to disinfect the hands of Austrian doctors, which Semmelweis noticed still carried the stench of decomposition from the dissection rooms to the patient examination rooms. Long before the germ theory of disease, Semmelweis theorized that "cadaveric particles" were transmitting decay from fresh medical cadavers to living patients, and he used the well-known "Labarraque's solutions" as the only known method to remove the smell of decay and tissue decomposition (which he found that soap did not). The solutions proved to be far more effective antiseptics than soap (Semmelweis was also aware of their greater efficacy, but not the reason), and this resulted in Semmelweis's celebrated success in stopping the transmission of childbed fever ("puerperal fever") in the maternity wards of Vienna General Hospital in Austria in 1847.[76]
Much later, during World War I in 1916, a standardized and diluted modification of Labarraque's solution containing hypochlorite (0.5%) and boric acid as an acidic stabilizer was developed by Henry Drysdale Dakin (who gave full credit to Labarraque's prior work in this area). Called Dakin's solution, the method of wound irrigation with chlorinated solutions allowed antiseptic treatment of a wide variety of open wounds, long before the modern antibiotic era. A modified version of this solution continues to be employed in wound irrigation in modern times, where it remains effective against bacteria that are resistant to multiple antibiotics (see Century Pharmaceuticals).[77]
Public sanitation

The first continuous application of chlorination to drinking U.S. water was installed in Jersey City, New Jersey in 1908.[78] By 1918, the US Department of Treasury called for all drinking water to be disinfected with chlorine. Chlorine is presently an important chemical for water purification (such as in water treatment plants), in disinfectants, and in bleach. Even small water supplies are now routinely chlorinated.[79]
Chlorine is usually used (in the form of hypochlorous acid) to kill bacteria and other microbes in drinking water supplies and public swimming pools. In most private swimming pools, chlorine itself is not used, but rather sodium hypochlorite, formed from chlorine and sodium hydroxide, or solid tablets of chlorinated isocyanurates. The drawback of using chlorine in swimming pools is that the chlorine reacts with the proteins in human hair and skin. The distinctive 'chlorine aroma' associated with swimming pools is not the result of chlorine itself, but of chloramine, a chemical compound produced by the reaction of free dissolved chlorine with amines in organic substances. As a disinfectant in water, chlorine is more than three times as effective against Escherichia coli as bromine, and more than six times as effective as iodine.[80] Increasingly, monochloramine itself is being directly added to drinking water for purposes of disinfection, a process known as chloramination.[81]
It is often impractical to store and use poisonous chlorine gas for water treatment, so alternative methods of adding chlorine are used. These include hypochlorite solutions, which gradually release chlorine into the water, and compounds like sodium dichloro-s-triazinetrione (dihydrate or anhydrous), sometimes referred to as "dichlor", and trichloro-s-triazinetrione, sometimes referred to as "trichlor". These compounds are stable while solid and may be used in powdered, granular, or tablet form. When added in small amounts to pool water or industrial water systems, the chlorine atoms hydrolyze from the rest of the molecule, forming hypochlorous acid (HOCl), which acts as a general biocide, killing germs, microorganisms, algae, and so on.[82][83]
Use as a weapon
World War I
Chlorine gas, also known as bertholite, was first used as a weapon in World War I by Germany on April 22, 1915 in the Second Battle of Ypres.[84][85] As described by the soldiers, it had the distinctive smell of a mixture of pepper and pineapple. It also tasted metallic and stung the back of the throat and chest. Chlorine reacts with water in the mucosa of the lungs to form hydrochloric acid, destructive to living tissue and potentially lethal. Human respiratory systems can be protected from chlorine gas by gas masks with activated charcoal or other filters, which makes chlorine gas much less lethal than other chemical weapons. It was pioneered by a German scientist later to be a Nobel laureate, Fritz Haber of the Kaiser Wilhelm Institute in Berlin, in collaboration with the German chemical conglomerate IG Farben, which developed methods for discharging chlorine gas against an entrenched enemy.[86] After its first use, both sides in the conflict used chlorine as a chemical weapon, but it was soon replaced by the more deadly phosgene and mustard gas.[87]
Iraq
Chlorine gas was also used during the Iraq War in Anbar Province in 2007, with insurgents packing truck bombs with mortar shells and chlorine tanks. The attacks killed two people from the explosives and sickened more than 350. Most of the deaths were caused by the force of the explosions rather than the effects of chlorine since the toxic gas is readily dispersed and diluted in the atmosphere by the blast. In some bombings, over a hundred civilians were hospitalized due to breathing difficulties. The Iraqi authorities tightened security for elemental chlorine, which is essential for providing safe drinking water to the population.[88][89]
On 24 October 2014, it was reported that the Islamic State of Iraq and the Levant had used chlorine gas in the town of Duluiyah, Iraq.[90] Laboratory analysis of clothing and soil samples confirmed the use of chlorine gas against Kurdish Peshmerga Forces in a vehicle-borne improvised explosive device attack on 23 January 2015 at the Highway 47 Kiske Junction near Mosul.[91]
Syria
The Syrian government has allegedly used chlorine as a chemical weapon[92] delivered from barrel bombs and rockets.[93][94]
Biological role
The chloride anion is an essential nutrient for metabolism. Chlorine is needed for the production of hydrochloric acid in the stomach and in cellular pump functions.[95] The main dietary source is table salt, or sodium chloride. Overly low or high concentrations of chloride in the blood are examples of electrolyte disturbances. Hypochloremia (having too little chloride) rarely occurs in the absence of other abnormalities. It is sometimes associated with hypoventilation.[96] It can be associated with chronic respiratory acidosis.[97] Hyperchloremia (having too much chloride) usually does not produce symptoms. When symptoms do occur, they tend to resemble those of hypernatremia (having too much sodium). Reduction in blood chloride leads to cerebral dehydration; symptoms are most often caused by rapid rehydration which results in cerebral edema. Hyperchloremia can affect oxygen transport.[98]
Hazards
| Hazards | |
|---|---|
| GHS pictograms |    |
| GHS Signal word | Danger |
GHS hazard statements |
H270, H315, H319, H331, H335, H400 |
| P220, P244, P261, P304, P340, P312, P403, P233, P410, P403[99] | |
| NFPA 704 (fire diamond) | |

Chlorine is a toxic gas that attacks the respiratory system, eyes, and skin.[101] Because it is denser than air, it tends to accumulate at the bottom of poorly ventilated spaces. Chlorine gas is a strong oxidizer, which may react with flammable materials.[102][103]
Chlorine is detectable with measuring devices in concentrations as low as 0.2 parts per million (ppm), and by smell at 3 ppm. Coughing and vomiting may occur at 30 ppm and lung damage at 60 ppm. About 1000 ppm can be fatal after a few deep breaths of the gas.[8] The IDLH (immediately dangerous to life and health) concentration is 10 ppm.[104] Breathing lower concentrations can aggravate the respiratory system and exposure to the gas can irritate the eyes.[105] The toxicity of chlorine comes from its oxidizing power. When chlorine is inhaled at concentrations greater than 30 ppm, it reacts with water and cellular fluid, producing hydrochloric acid (HCl) and hypochlorous acid (HClO).
When used at specified levels for water disinfection, the reaction of chlorine with water is not a major concern for human health. Other materials present in the water may generate disinfection by-products that are associated with negative effects on human health.[106][107]
In the United States, the Occupational Safety and Health Administration (OSHA) has set the permissible exposure limit for elemental chlorine at 1 ppm, or 3 mg/m3. The National Institute for Occupational Safety and Health has designated a recommended exposure limit of 0.5 ppm over 15 minutes.[104]
In the home, accidents occur when hypochlorite bleach solutions come into contact with certain acidic drain-cleaners to produce chlorine gas.[108] Hypochlorite bleach (a popular laundry additive) combined with ammonia (another popular laundry additive) produces chloramines, another toxic group of chemicals.[109]
Chlorine-induced cracking in structural materials

Chlorine is widely used for purifying water, especially potable water supplies and water used in swimming pools. Several catastrophic collapses of swimming pool ceilings have occurred from chlorine-induced stress corrosion cracking of stainless steel suspension rods.[110] Some polymers are also sensitive to attack, including acetal resin and polybutene. Both materials were used in hot and cold water domestic plumbing, and stress corrosion cracking caused widespread failures in the US in the 1980s and 1990s.[111]
Chlorine-iron fire
The element iron can combine with chlorine at high temperatures in a strong exothermic reaction, creating a chlorine-iron fire.[112][113] Chlorine-iron fires are a risk in chemical process plants, where much of the pipework that carries chlorine gas is made of steel.[112][113]
References
- Chlorine, Gas Encyclopaedia, Air Liquide
- Magnetic susceptibility of the elements and inorganic compounds, in Lide, D. R., ed. (2005). CRC Handbook of Chemistry and Physics (86th ed.). Boca Raton (FL): CRC Press. ISBN 0-8493-0486-5.
- Weast, Robert (1984). CRC, Handbook of Chemistry and Physics. Boca Raton, Florida: Chemical Rubber Company Publishing. pp. E110. ISBN 0-8493-0464-4.
- "The earliest salt production in the world: an early Neolithic exploitation in Poiana Slatinei-Lunca, Romania". Archived from the original on April 30, 2011. Retrieved 2008-07-10.
- Greenwood and Earnshaw, p. 789–92
- Scheele, Carl Wilhelm (1774). "Om Brunsten, eller Magnesia, och dess Egenskaper" [On braunstein [i.e., pyrolusite, manganese dioxide], or magnesia, and its properties]. Kongliga Vetenskaps Academiens Handlingar [Proceedings of the Royal Scientific Academy] (in Swedish). 35: 89–116, 177–194. In section 6 on pp. 93–94 of his paper, Scheele described how chlorine was produced when a mixture of hydrochloric acid and manganese dioxide (Brunsten) was heated: "6) (a) På 1/2 uns fint rifven Brunsten slogs 1 uns ren Spiritus salis. … samt lukten fo̊rsvunnen." ( 6) (a) On one half ounce of finely ground Braunstein [pyrolusite] was poured one ounce of pure spiritus salis [spirit of salt, hydrogen chloride]. After this mixture had been standing in the cold for one hour, the acid had assumed a dark brown colour. One part of this solution was poured into a glass, which was placed over the fire. The solution gave off an odour like warm aqua regia and after one quarter’s hour duration, it was as clear and colourless as water, and the smell had disappeared.) For an English translation of the relevant passages of this article, see: The Early History of Chlorine : Papers by Carl Wilhelm Scheele (1774), C. L. Berthollet (1785), Guyton de Morveau (1787), J. L. Gay-Lussac and L. J. Thenard (1809) (Edinburgh, Scotland: Alembic Club, 1912), pp. 5–10.
- "17 Chlorine". Elements.vanderkrogt.net. Archived from the original on 2010-01-23. Retrieved 2008-09-12.
- Greenwood and Earnshaw, pp. 792–93
- Ihde, Aaron John (1984). The development of modern chemistry. Courier Dover Publications. p. 158. ISBN 978-0-486-64235-2.
- Weeks, Mary Elvira (1932). "The discovery of the elements. XVII. The halogen family". Journal of Chemical Education. 9 (11): 1915. Bibcode:1932JChEd...9.1915W. doi:10.1021/ed009p1915.
- Gay-Lussac; Thenard (1809). "Extrait des mémoires lus à l'Institut national, depuis le 7 mars 1808 jusqu'au 27 février 1809" [Extracts from memoirs read at the national Institute, from 7 March 1808 to 27 February 1809]. Mémoires de Physique et de Chimie de la Société d'Arcueil. 2: 295–358. See: § De la nature et des propriétés de l'acide muriatique et de l'acide muriatique oxigéné (On the nature and properties of muriatic acid and of oxidized muriatic acid), pp. 339–358. From pp. 357–358: "Le gaz muriatique oxigéné n'est pas, en effect, décomposé … comme un corps composé." ("In fact, oxygenated muriatic acid is not decomposed by charcoal, and it might be supposed, from this fact and those that are communicated in this Memoir, that this gas is a simple body. The phenomena that it presents can be explained well enough on this hypothesis; we shall not seek to defend it, however, as it appears to us that they are still better explained by regarding oxygenated muriatic acid as a compound body.") For a full English translation of this section, see: Joseph Louis Gay-Lussac and Louis Jacques Thénard, "On the nature and the properties of muriatic acid and of oxygenated muriatic acid" (Lemoyne College, Syracuse, New York, USA)
- Davy, Humphry (1811). "The Bakerian Lecture. On some of the combinations of oxymuriatic gas and oxygene, and on the chemical relations of these principles, to inflammable bodies". Philosophical Transactions of the Royal Society of London. 101: 1–35. Bibcode:1811RSPT..101....1D. doi:10.1098/rstl.1811.0001. Davy named chlorine on p. 32: "After consulting some of the most eminent chemical philosophers in this country, it has been judged most proper to suggest a name founded upon one of its obvious and characteristic properties — its colour, and to call it Chlorine, or Chloric gas.* *From χλωρος."
- Schweigger, J.S.C. (1811). "Nachschreiben des Herausgebers, die neue Nomenclatur betreffend" [Postscript of the editor concerning the new nomenclature]. Journal für Chemie und Physik (in German). 3 (2): 249–255. On p. 251, Schweigger proposed the word "halogen": "Man sage dafür lieber mit richter Wortbildung Halogen (da schon in der Mineralogie durch Werner's Halit-Geschlecht dieses Wort nicht fremd ist) von αλς Salz und dem alten γενειν (dorisch γενεν) zeugen." (One should say instead, with proper morphology, "halogen" (this word is not strange since [it's] already in mineralogy via Werner's "halite" species) from αλς [als] "salt" and the old γενειν [genein] (Doric γενεν) "to beget".)
- In 1826, Berzelius coined the terms Saltbildare (salt-formers) and Corpora Halogenia (salt-making substances) for the elements chlorine, iodine, and fluorine. See: Berzelius, Jacob (1826). Årsberättelser om Framstegen i Physik och Chemie [Annual Report on Progress in Physics and Chemistry] (in Swedish). 6. Stockholm, Sweden: P.A. Norstedt & Söner. p. 187. From p. 187: "De förre af dessa, d. ä. de electronegativa, dela sig i tre klasser: 1) den första innehåller kroppar, som förenade med de electropositiva, omedelbart frambringa salter, hvilka jag derför kallar Saltbildare (Corpora Halogenia). Desse utgöras af chlor, iod och fluor *)." (The first of them [i.e., elements], i.e., the electronegative [ones], are divided into three classes: 1) The first includes substances which, [when] united with electropositive [elements], immediately produce salts, and which I therefore name "salt-formers" (salt-producing substances). These are chlorine, iodine, and fluorine *).)
- Snelders, H. A. M. (1971). "J. S. C. Schweigger: His Romanticism and His Crystal Electrical Theory of Matter". Isis. 62 (3): 328–338. doi:10.1086/350763. JSTOR 229946.
- Faraday, M. (1823). "On fluid chlorine". Philosophical Transactions of the Royal Society of London. 113: 160–164. Bibcode:1823RSPT..113..160F. doi:10.1098/rstl.1823.0016.
- Chodos, Alan (ed.). "This Month in Physics History September 4, 1821 and August 29, 1831: Faraday and Electromagnetism". American Physical Society. Archived from the original on June 15, 2010. Retrieved 2010-05-08.
- O'Connor J. J.; Robertson E. F. "Michael Faraday". School of Mathematics and Statistics, University of St Andrews, Scotland. Archived from the original on 2010-02-20. Retrieved 2010-05-08.
- "Bleaching". Encyclopædia Britannica (9th Edition (1875) and 10th Edition (1902) ed.). Archived from the original on 2012-05-24. Retrieved 2012-05-02.
- Aspin, Chris (1981). The Cotton Industry. Shire Publications Ltd. p. 24. ISBN 978-0-85263-545-2.
- Paul May. "Bleach (Sodium Hypochlorite)". University of Bristol. Archived from the original on 13 December 2016. Retrieved 13 December 2016.
- Greenwood and Earnshaw, p. 798
- Almqvist, Ebbe (2003). History of Industrial Gases. Springer Science & Business Media. p. 220. ISBN 978-0-306-47277-0.
- Bouvet, Maurice (1950). "Les grands pharmaciens: Labarraque (1777–1850)" [The great pharmacists: Labarraque (1777–1850)]. Revue d'Histoire de la Pharmacie (in French). 38 (128): 97–107. doi:10.3406/pharm.1950.8662.
- "Chlorine – History" (PDF). Archived from the original (PDF) on 21 February 2007. Retrieved 2008-07-10.
- "Weaponry: Use of Chlorine Gas Cylinders in World War I". historynet.com. 2006-06-12. Archived from the original on 2008-07-02. Retrieved 2008-07-10.
- Staff (29 July 2004). "On the Western Front, Ypres 1915". Veteran Affairs Canada. Archived from the original on 6 December 2008. Retrieved 2008-04-08.
- Lefebure, Victor; Wilson, Henry (2004). The Riddle of the Rhine: Chemical Strategy in Peace and War. Kessinger Publishing. ISBN 978-1-4179-3546-8.
- Greenwood and Earnshaw, pp. 800–4
- Greenwood and Earnshaw, pp. 804–09
- Cameron, A. G. W. (1973). "Abundance of the Elements in the Solar System" (PDF). Space Science Reviews. 15 (1): 121–46. Bibcode:1973SSRv...15..121C. doi:10.1007/BF00172440. Archived from the original (PDF) on 2011-10-21.
- Audi, Georges; Bersillon, Olivier; Blachot, Jean; Wapstra, Aaldert Hendrik (2003), "The NUBASE evaluation of nuclear and decay properties", Nuclear Physics A, 729: 3–128, Bibcode:2003NuPhA.729....3A, doi:10.1016/j.nuclphysa.2003.11.001
- M. Zreda; et al. (1991). "Cosmogenic chlorine-36 production rates in terrestrial rocks". Earth and Planetary Science Letters. 105 (1–3): 94–109. Bibcode:1991E&PSL.105...94Z. doi:10.1016/0012-821X(91)90123-Y.
- M. Sheppard and M. Herod (2012). "Variation in background concentrations and specific activities of 36Cl, 129I and U/Th-series radionuclides in surface waters". Journal of Environmental Radioactivity. 106: 27–34. doi:10.1016/j.jenvrad.2011.10.015. PMID 22304997.
- Greenwood and Earnshaw, pp. 853–56
- Greenwood and Earnshaw, pp. 809–12
- Greenwood and Earnshaw, pp. 812–16
- Greenwood and Earnshaw, pp. 818–19
- Greenwood and Earnshaw, pp. 821–44
- Greenwood and Earnshaw, pp. 842–44
- Greenwood and Earnshaw, pp. 824–8
- Greenwood and Earnshaw, pp. 835–42
- Greenwood and Earnshaw, pp. 828–31
- Greenwood and Earnshaw, pp. 832–35
- Greenwood and Earnshaw, pp. 875–80
- Greenwood and Earnshaw, pp. 844–50
- Greenwood and Earnshaw, pp. 883–5
- Greenwood and Earnshaw, pp. 856–70
- M. Rossberg et al. "Chlorinated Hydrocarbons" in Ullmann's Encyclopedia of Industrial Chemistry 2006, Wiley-VCH, Weinheim. doi:10.1002/14356007.a06_233.pub2
- Gordon W. Gribble (1998). "Naturally Occurring Organohalogen Compounds". Acc. Chem. Res. 31 (3): 141–52. doi:10.1021/ar9701777.
- Gordon W. Gribble (1999). "The diversity of naturally occurring organobromine compounds". Chemical Society Reviews. 28 (5): 335–46. doi:10.1039/a900201d.
- Kjeld C. Engvild (1986). "Chlorine-Containing Natural Compounds in Higher Plants". Phytochemistry. 25 (4): 7891–791. doi:10.1016/0031-9422(86)80002-4.
- Gribble, G. W. (1994). "The Natural production of chlorinated compounds". Environmental Science and Technology. 28 (7): 310A–319A. Bibcode:1994EnST...28..310G. doi:10.1021/es00056a712. PMID 22662801.
- Gribble, G. W. (1996). "Naturally occurring organohalogen compounds - A comprehensive survey". Progress in the Chemistry of Organic Natural Products. 68 (10): 1–423. doi:10.1021/np50088a001. PMID 8795309.
- Public Health Statement – Chloromethane Archived 2007-09-27 at the Wayback Machine, Centers for Disease Control, Agency for Toxic Substances and Disease Registry
- Connell, D.; et al. (1999). Introduction to Ecotoxicology. Blackwell Science. p. 68. ISBN 978-0-632-03852-7.
- Greenwood and Earnshaw, p. 795
- Holleman, Arnold Frederik; Wiberg, Egon (2001), Wiberg, Nils (ed.), Inorganic Chemistry, translated by Eagleson, Mary; Brewer, William, San Diego/Berlin: Academic Press/De Gruyter, p. 408, ISBN 0-12-352651-5
- "The diaphragm cell process". Euro Chlor. Archived from the original on 2011-11-11. Retrieved 2007-08-15.
- "The membrane cell process". Euro Chlor. Archived from the original on 2011-11-11. Retrieved 2007-08-15.
- Schmittinger, Peter et al. (2006) "Chlorine" in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co., doi:10.1002/14356007.a06_399.pub2
- Greenwood and Earnshaw, pp. 796–800
- Greenwood 1997, p. 798.
- Greenwood 1997, p. 793.
- Hoefer, Jean Chrétien Ferdinand (ed.). "Labarraque, Antoine-Germain". Nouvelle biographie universelle. 28. pp. 323–24. OL 24229911M.
- Knight, Charles (1867). Arts and sciences. 1. Bradbury, Evans & Co. p. 427.
- Bouvet, Maurice (1950). "Les grands pharmaciens: Labarraque (1777–1850)" [The great pharmacists: Labarraque (1777–1850)]. Revue d'Histoire de la Pharmacie (in French). 38 (128): 97–107. doi:10.3406/pharm.1950.8662.
- Gédéon, Andras (2006). Science and technology in medicine. Springer. pp. 181–82. ISBN 978-0-387-27874-2. Archived from the original on 2015-12-31.
- Labarraque, Antoine Germain (1828). On the disinfecting properties of Labarraque's preparations of chlorine. Translated by James Scott. p. 8. Archived from the original on 2015-12-31.
- Scott, James (trans.). On the disinfecting properties of Labarraque's preparations of chlorine Archived 2015-12-31 at the Wayback Machine (S. Highley, 1828) Accessed Nov 1, 2011.
- Corbin, Alain (1988). The Foul and the Fragrant: Odor and the French Social Imagination Archived 2015-12-31 at the Wayback Machine. Harvard University Press. pp. 121–22.
- Lewis, Kenneth A. (2010). "Ch. 9 Hypochlorination – Sodium Hypochlorite" (PDF). White's Handbook of Chlorination and Alternative Disinfectants. Hoboken, NJ: Wiley. p. 452. doi:10.1002/9780470561331.ch9. ISBN 978-0-470-56133-1.
- Vinten-Johansen, Peter, Howard Brody, Nigel Paneth, Stephen Rachman and Michael Rip. (2003). Cholera, Chloroform, and the Science of Medicine. New York:Oxford University.
- Hemphill, Sandra. (2007). The Strange Case of the Broad Street Pump: John Snow and the Mystery of Cholera. Los Angeles:University of California
- Johnson, Steven. (2006). The Ghost Map: The Story of London's Most Terrifying Epidemic and How It Changed Science, Cities, and the Modern World. New York :Riverhead Books
- "Chlorine Story". americanchemistry. Archived from the original on 2011-04-29. Retrieved 2008-07-10.CS1 maint: BOT: original-url status unknown (link)
- Rezayat, C.; Widmann, W. D.; Hardy, M. A. (2006). "Henry Drysdale Dakin: More Than His Solution". Current Surgery. 63 (3): 194–96. doi:10.1016/j.cursur.2006.04.009. PMID 16757372.
- Joseph Cotruvo, Victor Kimm, Arden Calvert. “Drinking Water: A Half Century of Progress.” EPA Alumni Association. March 1, 2016.
- Hammond, C. R. (2000). The Elements, in Handbook of Chemistry and Physics (81st ed.). CRC press. ISBN 978-0-8493-0481-1.
- Koski T. A.; Stuart L. S.; Ortenzio L. F. (1966). "Comparison of chlorine, bromine, iodine as disinfectants for swimming pool water". Applied Microbiology. 14 (2): 276–79. doi:10.1128/AEM.14.2.276-279.1966. PMC 546668. PMID 4959984.
- "Disinfection with chloramine". Centers for Disease Control and Prevention (CDC). Atlanta, Georgia, USA. Archived from the original on 2019-01-20. Retrieved 2019-01-20.
- Greenwood 1997, p. 860.
- Wiberg 2001, p. 411.
- "Battle of Ypres" The Canadian Encyclopedia
- Everts, Sarah (February 23, 2015). "When Chemicals Became Weapons of War". Chemical & Engineering News. 93 (8). Archived from the original on March 30, 2016.
- Smil, Vaclav (2004-04-01). Enriching the Earth: Fritz Haber, Carl Bosch, and the Transformation of World Food Production. p. 226. ISBN 978-0-262-69313-4. Archived from the original on 2015-12-31.
- "Weapons of War: Poison Gas". First World War.com. Archived from the original on 2007-08-21. Retrieved 2007-08-12.
- Mahdi, Basim (2007-03-17). "Iraq gas attack makes hundreds ill". CNN. Archived from the original on 2007-03-17. Retrieved 2007-03-17.
- "'Chlorine bomb' hits Iraq village". BBC News. 2007-05-17. Archived from the original on 2007-05-26. Retrieved 2007-05-17.
- "ISIS chlorine attack on security forces confirmed – Iraq officials". RT. October 24, 2014. Archived from the original on October 24, 2014.
- "Lab report on chlorine gas usage" (PDF). Kurdistan Region Security Council. March 14, 2015.
- Gladstone, Rick (2017-02-13). "Syria Used Chlorine Bombs Systematically in Aleppo, Report Says". The New York Times. Archived from the original on 2017-05-15. Retrieved 2017-05-10.
- "Syrian forces 'drop chlorine' on Aleppo". BBC News. 2016-09-07. Archived from the original on 2017-05-13. Retrieved 2017-05-10.
- "Ignoring UN, Russia and Assad continue Syrian chemical weapons and bombing attacks labeled war crimes". 2017-03-06. Archived from the original on 2017-04-25. Retrieved 2017-05-11.
- "Blood (Serum) Chloride Level Test". Archived from the original on 31 March 2009. Retrieved 30 April 2010.
- Lavie, CJ; Crocker, EF; Key, KJ; Ferguson, TG (October 1986). "Marked hypochloremic metabolic alkalosis with severe compensatory hypoventilation". South. Med. J. 79 (10): 1296–99. doi:10.1097/00007611-198610000-00025. PMID 3764530.
- Levitin, H; Branscome, W; Epstein, FH (December 1958). "The pathogenesis of hypochloremia in respiratory acidosis". J. Clin. Invest. 37 (12): 1667–75. doi:10.1172/JCI103758. PMC 1062852. PMID 13611033.
- Cambier, C; Detry, B; Beerens, D; et al. (October 1998). "Effects of hyperchloremia on blood oxygen binding in healthy calves". J. Appl. Physiol. 85 (4): 1267–72. doi:10.1152/jappl.1998.85.4.1267. PMID 9760315.
- "Chlorine 295132".
- "Msds - 295132".
- "Facts About Chlorine". www.bt.cdc.gov. Archived from the original on 2016-04-23. Retrieved 2016-04-12.CS1 maint: BOT: original-url status unknown (link)
- "Chlorine MSDS" (PDF). 1997-10-23. Archived from the original (PDF) on 2007-09-26.
- NOAA Office of Response and Restoration, US GOV. "Chlorine". noaa.gov. Archived from the original on 15 October 2015. Retrieved 25 August 2015.
- NIOSH Pocket Guide to Chemical Hazards. "#0115". National Institute for Occupational Safety and Health (NIOSH).
- Winder, Chris (2001). "The Toxicology of Chlorine". Environmental Research. 85 (2): 105–14. Bibcode:2001ER.....85..105W. doi:10.1006/enrs.2000.4110. PMID 11161660.
- "What's in your Water?: Disinfectants Create Toxic By-products". ACES News. College of Agricultural, Consumer and Environmental Sciences – University of Illinois at Urbana-Champaign. 2009-03-31. Archived from the original on 2014-09-03. Retrieved 2009-03-31.
- Richardson, Susan D.; Plewa, Michael J.; Wagner, Elizabeth D.; Schoeny, Rita; DeMarini, David M. (2007). "Occurrence, genotoxicity, and carcinogenicity of regulated and emerging disinfection by-products in drinking water: A review and roadmap for research". Mutation Research/Reviews in Mutation Research. 636 (1–3): 178–242. doi:10.1016/j.mrrev.2007.09.001. PMID 17980649.
- Berezow, Alex. "Why You Should Never Mix Different Drain Cleaners". Forbes. Archived from the original on 2016-04-25. Retrieved 2016-04-12.
- "Bleach Mixing Dangers : Washington State Dept. of Health". www.doh.wa.gov. Archived from the original on 2016-04-14. Retrieved 2016-04-12.
- Bertolini, Luca; Elsener, Bernhard; Pedeferri, Pietro; Polder, Rob B. (2004). Corrosion of steel in concrete: prevention, diagnosis, repair. Wiley-VCH. p. 148. ISBN 978-3-527-30800-2.
- Lewis, P.R. (1 January 2000). Polymer Product Failure. iSmithers Rapra Publishing. pp. 19–. ISBN 978-1-85957-192-7. Archived from the original on 10 May 2013. Retrieved 2011-04-30.
- "Chlorine: Product Datasheet" (PDF). Bayer MaterialScience AG. 2008-04-21. Archived from the original (PDF) on September 15, 2012. Retrieved 2013-12-17.
- Sanders, Roy E. (2004). Chemical Process Safety: Learning from Case Histories, 3rd Revised edition. Oxford: Elsevier Science & Technology. p. 92. ISBN 978-0-7506-7749-3.
Notes
- van Helmont, Joannis Baptistae (1682). Opera omnia [All Works] (in Latin). Frankfurt-am-Main, (Germany): Johann Just Erythropel. From "Complexionum atque mistionum elementalium figmentum." (Formation of combinations and of mixtures of elements), §37, p. 105: "Accipe salis petrae, vitrioli, & alumnis partes aequas: exsiccato singula, & connexis simul, distilla aquam. Quae nil aliud est, quam merum sal volatile. Hujus accipe uncias quatuor, salis armeniaci unciam junge, in forti vitro, alembico, per caementum (ex cera, colophonia, & vitri pulverre) calidissime affusum, firmato; mox, etiam in frigore, Gas excitatur, & vas, utut forte, dissilit cum fragore." (Take equal parts of saltpeter [i.e., sodium nitrate], vitriol [i.e., concentrated sulfuric acid], and alum: dry each and combine simultaneously; distill off the water [i.e., liquid]. That [distillate] is nothing else than pure volatile salt [i.e., spirit of nitre, nitric acid]. Take four ounces of this [viz, nitric acid], add one ounce of Armenian salt [i.e., ammonium chloride], [place it] in a strong glass alembic sealed by cement ([made] from wax, rosin, and powdered glass) [that has been] poured very hot; soon, even in the cold, gas is stimulated, and the vessel, however strong, bursts into fragments.) From "De Flatibus" (On gases), p. 408: "Sal armeniacus enim, & aqua chrysulca, quae singula per se distillari, possunt, & pati calorem: sin autem jungantur, & intepescant, non possunt non, quin statim in Gas sylvestre, sive incoercibilem flatum transmutentur." (Truly Armenian salt [i.e., ammonium chloride] and nitric acid, each of which can be distilled by itself, and submitted to heat; but if, on the other hand, they be combined and become warm, they cannot but be changed immediately into carbon dioxide [note: van Helmont’s identification of the gas is mistaken] or an incondensable gas.)
See also:- Helmont, Johannes (Joan) Baptista Van, Encyclopedia.Com: "Others were chlorine gas from the reaction of nitric acid and sal ammoniac; … "
- Wisniak, Jaime (2009) "Carl Wilhelm Scheele," Revista CENIC Ciencias Químicas, 40 (3): 165–173 ; see p. 168: "Early in the seventeenth century Johannes Baptiste van Helmont (1579-1644) mentioned that when sal marin (sodium chloride) or sal ammoniacus and aqua chrysulca (nitric acid) were mixed together, a flatus incoercible (non-condensable gas) was evolved."
Bibliography
- Greenwood, Norman N.; Earnshaw, Alan (1997). Chemistry of the Elements (2nd ed.). Butterworth-Heinemann. ISBN 978-0-08-037941-8.
External links
| Wikimedia Commons has media related to Chlorine. |
- Chlorine at The Periodic Table of Videos (University of Nottingham)
- Agency for Toxic Substances and Disease Registry: Chlorine
- Electrolytic production
- Production and liquefaction of chlorine
- Chlorine Production Using Mercury, Environmental Considerations and Alternatives
- National Pollutant Inventory – Chlorine
- National Institute for Occupational Safety and Health – Chlorine Page
- Chlorine Institute – Trade association representing the chlorine industry
- Chlorine Online – the web portal of Eurochlor – the business association of the European chlor-alkali industry
- . Encyclopædia Britannica. 6 (11th ed.). 1911. pp. 254–56.
| Authority control |
|
|---|