Tumor necrosis factor
Tumor necrosis factor (TNF, cachexin, or cachectin; once named as tumor necrosis factor alpha or TNFα) is a cell signaling protein (cytokine) involved in systemic inflammation and is one of the cytokines that make up the acute phase reaction. It is produced chiefly by activated macrophages, although it can be produced by many other cell types such as T helper cells, natural killer cells, neutrophils, mast cells, eosinophils, and neurons.[5] TNF is a member of the TNF superfamily, consisting of various transmembrane proteins with a homologous TNF domain.



The primary role of TNF is in the regulation of immune cells. TNF, being an endogenous pyrogen, is able to induce fever, apoptotic cell death, cachexia, inflammation and to inhibit tumorigenesis, viral replication, and respond to sepsis via IL-1 and IL-6-producing cells. Dysregulation of TNF production has been implicated in a variety of human diseases including Alzheimer's disease,[6] cancer,[7] major depression,[8] psoriasis[9] and inflammatory bowel disease (IBD).[10] Though controversial, studies of depression and IBD have been linked to increased levels of TNF.[11][12] Recombinant TNF is used as an immunostimulant under the INN tasonermin. TNF can be produced ectopically in the setting of malignancy and parallels parathyroid hormone both in causing secondary hypercalcemia and in the cancers with which excessive production is associated.
Discovery
The theory of an anti-tumoral response of the immune system in vivo was recognized by the physician William B. Coley. In 1968, Gale A Granger from the University of California, Irvine, reported a cytotoxic factor produced by lymphocytes and named it lymphotoxin (LT).[13] Credit for this discovery is shared by Nancy H. Ruddle from Yale University, who reported the same activity in a series of back-to-back articles published in the same month.[14] Subsequently, in 1975 Lloyd J. Old from Memorial Sloan-Kettering Cancer Center, New York, reported another cytotoxic factor produced by macrophages and named it tumor necrosis factor (TNF).[15] Both factors were described based on their ability to kill mouse fibrosarcoma L-929 cells. These concepts were extended to systemic disease in 1981, when Ian A. Clark, from the Australian National University, in collaboration with Elizabeth Carswell in Old's group, working with pre-sequencing era data, reasoned that excessive production of TNF causes malaria disease and endotoxin poisoning.[16][17]
The cDNAs encoding LT and TNF were cloned in 1984[18] and were revealed to be similar. The binding of TNF to its receptor and its displacement by LT confirmed the functional homology between the two factors. The sequential and functional homology of TNF and LT led to the renaming of TNF as TNFα and LT as TNFβ. In 1985, Bruce A. Beutler and Anthony Cerami discovered that cachectin (a hormone which induces cachexia) was actually TNF.[19] They then identified TNF as a mediator of lethal endotoxin poisoning.[20] Kevin J. Tracey and Cerami discovered the key mediator role of TNF in lethal septic shock, and identified the therapeutic effects of monoclonal anti-TNF antibodies.[21][22] More recently, research in the Laboratory of Mark Mattson has shown that TNF can prevent the death/apoptosis of neurons by a mechanism involving activation of the transcription factor NF-kappaB which induces the expression of Mn-SOD and Bcl-2.
Gene
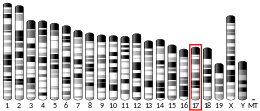
The human TNF gene was cloned in 1985.[23] It maps to chromosome 6p21.3, spans about 3 kilobases and contains 4 exons. The last exon shares similarity with lymphotoxin alpha (LTA, once named as TNF-β).[24] The three prime untranslated region ( 3'-UTR) of TNF contains an AU-rich element (ARE).
Structure
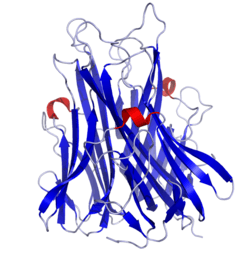
TNF is primarily produced as a 233-amino acid-long type II transmembrane protein arranged in stable homotrimers.[25][26] From this membrane-integrated form the soluble homotrimeric cytokine (sTNF) is released via proteolytic cleavage by the metalloprotease TNF alpha converting enzyme (TACE, also called ADAM17).[27] The soluble 51 kDa trimeric sTNF tends to dissociate at concentrations below the nanomolar range, thereby losing its bioactivity. The secreted form of human TNF takes on a triangular pyramid shape, and weighs around 17-kDa. Both the secreted and the membrane bound forms are biologically active, although the specific functions of each is controversial. But, both forms do have overlapping and distinct biological activities.[28]
The common house mouse TNF and human TNF are structurally different.[29] The 17-kilodalton (kDa) TNF protomers (185-amino acid-long) are composed of two antiparallel β-pleated sheets with antiparallel β-strands, forming a 'jelly roll' β-structure, typical for the TNF family, but also found in viral capsid proteins.
Cell signaling
TNF can bind two receptors, TNFR1 (TNF receptor type 1; CD120a; p55/60) and TNFR2 (TNF receptor type 2; CD120b; p75/80). TNFR1 is 55-kDa and TNFR2 is 75-kDa.[30] TNFR1 is expressed in most tissues, and can be fully activated by both the membrane-bound and soluble trimeric forms of TNF, whereas TNFR2 is found typically in cells of the immune system, and responds to the membrane-bound form of the TNF homotrimer. As most information regarding TNF signaling is derived from TNFR1, the role of TNFR2 is likely underestimated.
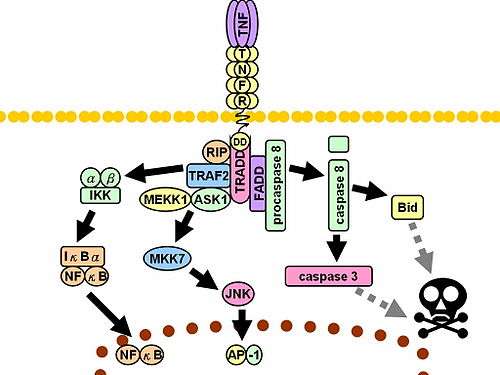
Upon contact with their ligand, TNF receptors also form trimers, their tips fitting into the grooves formed between TNF monomers. This binding causes a conformational change to occur in the receptor, leading to the dissociation of the inhibitory protein SODD from the intracellular death domain. This dissociation enables the adaptor protein TRADD to bind to the death domain, serving as a platform for subsequent protein binding. Following TRADD binding, three pathways can be initiated.[31][32]
- Activation of NF-κB: TRADD recruits TRAF2 and RIP. TRAF2 in turn recruits the multicomponent protein kinase IKK, enabling the serine-threonine kinase RIP to activate it. An inhibitory protein, IκBα, that normally binds to NF-κB and inhibits its translocation, is phosphorylated by IKK and subsequently degraded, releasing NF-κB. NF-κB is a heterodimeric transcription factor that translocates to the nucleus and mediates the transcription of a vast array of proteins involved in cell survival and proliferation, inflammatory response, and anti-apoptotic factors.
- Activation of the MAPK pathways: Of the three major MAPK cascades, TNF induces a strong activation of the stress-related JNK group, evokes moderate response of the p38-MAPK, and is responsible for minimal activation of the classical ERKs. TRAF2/Rac activates the JNK-inducing upstream kinases of MLK2/MLK3,[33] TAK1, MEKK1 and ASK1 (either directly or through GCKs and Trx, respectively). SRC- Vav- Rac axis activates MLK2/MLK3 and these kinases phosphorylate MKK7, which then activates JNK. JNK translocates to the nucleus and activates transcription factors such as c-Jun and ATF2. The JNK pathway is involved in cell differentiation, proliferation, and is generally pro-apoptotic.
- Induction of death signaling: Like all death-domain-containing members of the TNFR superfamily, TNFR1 is involved in death signaling.[34] However, TNF-induced cell death plays only a minor role compared to its overwhelming functions in the inflammatory process. Its death-inducing capability is weak compared to other family members (such as Fas), and often masked by the anti-apoptotic effects of NF-κB. Nevertheless, TRADD binds FADD, which then recruits the cysteine protease caspase-8. A high concentration of caspase-8 induces its autoproteolytic activation and subsequent cleaving of effector caspases, leading to cell apoptosis.
The myriad and often-conflicting effects mediated by the above pathways indicate the existence of extensive cross-talk. For instance, NF-κB enhances the transcription of C-FLIP, Bcl-2, and cIAP1 / cIAP2, inhibitory proteins that interfere with death signaling. On the other hand, activated caspases cleave several components of the NF-κB pathway, including RIP, IKK, and the subunits of NF-κB itself. Other factors, such as cell type, concurrent stimulation of other cytokines, or the amount of reactive oxygen species (ROS) can shift the balance in favor of one pathway or another. Such complicated signaling ensures that, whenever TNF is released, various cells with vastly diverse functions and conditions can all respond appropriately to inflammation.Both protein molecules tumor necrosis factor alpha and keratin 17 appear to be related in case of oral submucous fibrosis<refGhada A. Abd El Latif, Tumor necrosis factor alpha and keratin 17 expression in oral submucous fibrosis in rat model, E.D.J. Vol. 65, (1) Pp 277-288; 2019. DOI: 10.21608/edj.2015.71414></ref>
In animal models TNF selectively kills autoreactive T cells.[35]
Enzyme regulation
This protein may use the morpheein model of allosteric regulation.[36]
Physiology
TNF was thought to be produced primarily by macrophages,[37] but it is produced also by a broad variety of cell types including lymphoid cells, mast cells, endothelial cells, cardiac myocytes, adipose tissue, fibroblasts, and neurons.[5] Large amounts of TNF are released in response to lipopolysaccharide, other bacterial products, and Interleukin-1 (IL-1). In the skin, mast cells appear to be the predominant source of pre-formed TNF, which can be released upon inflammatory stimulus (e.g., LPS).[38]
It has a number of actions on various organ systems, generally together with IL-1 and Interleukin-6 (IL-6):
- On the hypothalamus:
- On the liver: stimulating the acute phase response, leading to an increase in C-reactive protein and a number of other mediators. It also induces insulin resistance by promoting serine-phosphorylation of insulin receptor substrate-1 (IRS-1), which impairs insulin signaling
- It is a potent chemoattractant for neutrophils, and promotes the expression of adhesion molecules on endothelial cells, helping neutrophils migrate.
- On macrophages: stimulates phagocytosis, and production of IL-1 oxidants and the inflammatory lipid Prostaglandin E2 (PGE2)
- On other tissues: increasing insulin resistance. TNF phosphorylates insulin receptor serine residues, blocking signal transduction.
- On metabolism and food intake: regulates bitter taste perception.[39]
A local increase in concentration of TNF will cause the cardinal signs of Inflammation to occur: heat, swelling, redness, pain and loss of function.
Whereas high concentrations of TNF induce shock-like symptoms, the prolonged exposure to low concentrations of TNF can result in cachexia, a wasting syndrome. This can be found, for example, in cancer patients.
Said et al. showed that TNF causes an IL-10-dependent inhibition of CD4 T-cell expansion and function by up-regulating PD-1 levels on monocytes which leads to IL-10 production by monocytes after binding of PD-1 by PD-L.[40]
The research of Pedersen et al. indicates that TNF increase in response to sepsis is inhibited by the exercise-induced production of myokines. To study whether acute exercise induces a true anti-inflammatory response, a model of ‘low grade inflammation’ was established in which a low dose of E. coli endotoxin was administered to healthy volunteers, who had been randomised to either rest or exercise prior to endotoxin administration. In resting subjects, endotoxin induced a 2- to 3-fold increase in circulating levels of TNF. In contrast, when the subjects performed 3 hours of ergometer cycling and received the endotoxin bolus at 2.5 h, the TNF response was totally blunted.[41] This study provides some evidence that acute exercise may inhibit TNF production.[42]
Pharmacology
TNF promotes the inflammatory response, which, in turn, causes many of the clinical problems associated with autoimmune disorders such as rheumatoid arthritis, ankylosing spondylitis, inflammatory bowel disease, psoriasis, hidradenitis suppurativa and refractory asthma. These disorders are sometimes treated by using a TNF inhibitor. This inhibition can be achieved with a monoclonal antibody such as infliximab (Remicade) binding directly to TNF, adalimumab (Humira), certolizumab pegol (Cimzia) or with a decoy circulating receptor fusion protein such as etanercept (Enbrel) which binds to TNF with greater affinity than the TNFR.[43]
On the other hand some patients treated with TNF inhibitors develop an aggravation of their disease or new onset of autoimmunity. TNF seems to have an immunosuppressive facet as well. One explanation for a possible mechanism is this observation that TNF has a positive effect on regulatory T cells (Tregs), due to its binding to the tumor necrosis factor receptor 2 (TNFR2).[44]
Anti-TNF therapy has shown only modest effects in cancer therapy. Treatment of renal cell carcinoma with infliximab resulted in prolonged disease stabilization in certain patients. Etanercept was tested for treating patients with breast cancer and ovarian cancer showing prolonged disease stabilization in certain patients via downregulation of IL-6 and CCL2. On the other hand, adding infliximab or etanercept to gemcitabine for treating patients with advanced pancreatic cancer was not associated with differences in efficacy when compared with placebo.[45]
Nomenclature
Because LTα is no longer referred to as TNFβ [48], TNFα, as the previous gene symbol, is now simply called TNF, as shown in HGNC (HUGO Gene Nomenclature Committee) database.
References
- ENSG00000230108, ENSG00000223952, ENSG00000204490, ENSG00000228321, ENSG00000232810, ENSG00000228849, ENSG00000206439 GRCh38: Ensembl release 89: ENSG00000228978, ENSG00000230108, ENSG00000223952, ENSG00000204490, ENSG00000228321, ENSG00000232810, ENSG00000228849, ENSG00000206439 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000024401 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- 9218250
- Swardfager W, Lanctôt K, Rothenburg L, Wong A, Cappell J, Herrmann N (2010). "A meta-analysis of cytokines in Alzheimer's disease". Biol Psychiatry. 68 (10): 930–941. doi:10.1016/j.biopsych.2010.06.012. PMID 20692646.
- Locksley RM, Killeen N, Lenardo MJ (2001). "The TNF and TNF receptor superfamilies: integrating mammalian biology". Cell. 104 (4): 487–501. doi:10.1016/S0092-8674(01)00237-9. PMID 11239407.
- Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctôt KL (2010). "A meta-analysis of cytokines in major depression". Biol Psychiatry. 67 (5): 446–457. doi:10.1016/j.biopsych.2009.09.033. PMID 20015486.
- Victor FC, Gottlieb AB (2002). "TNF-alpha and apoptosis: implications for the pathogenesis and treatment of psoriasis". J Drugs Dermatol. 1 (3): 264–75. PMID 12851985.
- Brynskov J, Foegh P, Pedersen G, Ellervik C, Kirkegaard T, Bingham A, Saermark T (2002). "Tumour necrosis factor alpha converting enzyme (TACE) activity in the colonic mucosa of patients with inflammatory bowel disease". Gut. 51 (1): 37–43. doi:10.1136/gut.51.1.37. PMC 1773288. PMID 12077089.
- Mikocka-Walus AA, Turnbull DA, Moulding NT, Wilson IG, Andrews JM, Holtmann GJ (2007). "Controversies surrounding the comorbidity of depression and anxiety in inflammatory bowel disease patients: a literature review". Inflammatory Bowel Diseases. 13 (2): 225–234. doi:10.1002/ibd.20062. PMID 17206706.
- Bobińska K, Gałecka E, Szemraj J, Gałecki P, Talarowska M (2017). "Is there a link between TNF gene expression and cognitive deficits in depression?". Acta Biochim. Pol. 64 (1): 65–73. doi:10.18388/abp.2016_1276. PMID 27991935.
- Kolb WP, Granger GA (1968). "Lymphocyte in vitro cytotoxicity: characterization of human lymphotoxin". Proc. Natl. Acad. Sci. U.S.A. 61 (4): 1250–5. Bibcode:1968PNAS...61.1250K. doi:10.1073/pnas.61.4.1250. PMC 225248. PMID 5249808.
- Ruddle NH, Waksman BH (December 1968). "Cytotoxicity mediated by soluble antigen and lymphocytes in delayed hypersensitivity. 3. Analysis of mechanism". J. Exp. Med. 128 (6): 1267–79. doi:10.1084/jem.128.6.1267. PMC 2138574. PMID 5693925.
- Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B (1975). "An endotoxin-induced serum factor that causes necrosis of tumors". Proc. Natl. Acad. Sci. U.S.A. 72 (9): 3666–70. Bibcode:1975PNAS...72.3666C. doi:10.1073/pnas.72.9.3666. PMC 433057. PMID 1103152.
- Clark IA, Virelizier JL, Carswell EA, Wood PR (June 1981). "Possible importance of macrophage-derived mediators in acute malaria". Infect. Immun. 32 (3): 1058–66. PMC 351558. PMID 6166564.
- Clark IA (July 1982). "Suggested importance of monokines in pathophysiology of endotoxin shock and malaria". Klin. Wochenschr. 60 (14): 756–8. doi:10.1007/BF01716573. PMID 6181289.
- Pennica D, Nedwin GE, Hayflick JS, Seeburg PH, Derynck R, Palladino MA, Kohr WJ, Aggarwal BB, Goeddel DV (1984). "Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin". Nature. 312 (5996): 724–9. Bibcode:1984Natur.312..724P. doi:10.1038/312724a0. PMID 6392892.
- Beutler B, Greenwald D, Hulmes JD, Chang M, Pan YC, Mathison J, Ulevitch R, Cerami A (1985). "Identity of tumour necrosis factor and the macrophage-secreted factor cachectin". Nature. 316 (6028): 552–4. Bibcode:1985Natur.316..552B. doi:10.1038/316552a0. PMID 2993897.
- Beutler B, Milsark IW, Cerami AC (August 1985). "Passive immunization against cachectin/tumor necrosis factor protects mice from lethal effect of endotoxin". Science. 229 (4716): 869–71. Bibcode:1985Sci...229..869B. doi:10.1126/science.3895437. PMID 3895437.
- Tracey KJ, Beutler B, Lowry SF, Merryweather J, Wolpe S, Milsark IW, Hariri RJ, Fahey TJ, Zentella A, Albert JD (October 1986). "Shock and tissue injury induced by recombinant human cachectin". Science. 234 (4775): 470–74. Bibcode:1986Sci...234..470T. doi:10.1126/science.3764421. PMID 3764421.
- Tracey KJ, Fong Y, Hesse DG, Manogue KR, Lee AT, Kuo GC, Lowry SF, Cerami A (December 1987). "Anti-cachectin/TNF monoclonal antibodies prevent septic shock during lethal bacteraemia". Nature. 330 (6149): 662–64. Bibcode:1987Natur.330..662T. doi:10.1038/330662a0. PMID 3317066.
- Old LJ (1985). "Tumor necrosis factor (TNF)". Science. 230 (4726): 630–2. Bibcode:1985Sci...230..630O. doi:10.1126/science.2413547. PMID 2413547.
- Nedwin GE, Naylor SL, Sakaguchi AY, Smith D, Jarrett-Nedwin J, Pennica D, Goeddel DV, Gray PW (1985). "Human lymphotoxin and tumor necrosis factor genes: structure, homology and chromosomal localization". Nucleic Acids Res. 13 (17): 6361–73. doi:10.1093/nar/13.17.6361. PMC 321958. PMID 2995927.
- Kriegler M, Perez C, DeFay K, Albert I, Lu SD (1988). "A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: ramifications for the complex physiology of TNF". Cell. 53 (1): 45–53. doi:10.1016/0092-8674(88)90486-2. PMID 3349526.
- Tang P, Klostergaard J (1996). "Human pro-tumor necrosis factor is a homotrimer". Biochemistry. 35 (25): 8216–25. doi:10.1021/bi952182t. PMID 8679576.
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP (1997). "A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells". Nature. 385 (6618): 729–33. Bibcode:1997Natur.385..729B. doi:10.1038/385729a0. PMID 9034190.
- Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL (September 2003). "Anti-TNF-α therapies: the next generation". Nature Reviews Drug Discovery. 2 (9): 736–46. Bibcode:2003Natur.002..736B. doi:10.1038/nrd1175. PMID 12951580.
- Olszewski MB, Groot AJ, Dastych J, Knol EF (May 2007). "TNF trafficking to human mast cell granules: mature chain-dependent endocytosis". J. Immunol. 178 (9): 5701–9. doi:10.4049/jimmunol.178.9.5701. PMID 17442953.
In human cells, contrary to results previously obtained in a rodent model, TNF seems not to be glycosylated and, thus, trafficking is carbohydrate independent. In an effort to localize the amino acid motif responsible for granule targeting, we constructed additional fusion proteins and analyzed their trafficking, concluding that granule-targeting sequences are localized in the mature chain of TNF and that the cytoplasmic tail is expendable for endocytotic sorting of this cytokine, thus excluding direct interactions with intracellular adaptor proteins
- Theiss. A. L. et al. 2005. Tumor necrosis factor (TNF) alpha increases collagen accumulation and proliferation in intestinal myofibrobasts via TNF Receptor 2. The Journal of Biological Chemistry. [Online] 2005. Available at: http://www.jbc.org/content/280/43/36099.long Accessed: 21/10/14
- Wajant H, Pfizenmaier K, Scheurich P (2003). "Tumor necrosis factor signaling". Cell Death Differ. 10 (1): 45–65. doi:10.1038/sj.cdd.4401189. PMID 12655295.
- Chen G, Goeddel DV (2002). "TNF-R1 signaling: a beautiful pathway". Science. 296 (5573): 1634–5. Bibcode:2002Sci...296.1634C. doi:10.1126/science.1071924. PMID 12040173.
- Kant S, Swat W, Zhang S, Zhang ZY, Neel BG, Flavell RA, Davis RJ (2011). "TNF-stimulated MAP kinase activation mediated by a Rho family GTPase signaling pathway". Genes Dev. 25 (19): 2069–78. doi:10.1101/gad.17224711. PMC 3197205. PMID 21979919.
- Gaur U, Aggarwal BB (2003). "Regulation of proliferation, survival and apoptosis by members of the TNF superfamily". Biochem. Pharmacol. 66 (8): 1403–8. doi:10.1016/S0006-2952(03)00490-8. PMID 14555214.
- Ban L, Zhang J, Wang L, Kuhtreiber W, Burger D, Faustman DL (2008). "Selective death of autoreactive T cells in human diabetes by TNF or TNF receptor 2 agonism". PNAS. 105 (36): 13644–13649. doi:10.1073/pnas.0803429105. PMC 2533243. PMID 18755894.
- Selwood T, Jaffe EK (2011). "Dynamic dissociating homo-oligomers and the control of protein function". Arch. Biochem. Biophys. 519 (2): 131–43. doi:10.1016/j.abb.2011.11.020. PMC 3298769. PMID 22182754.
- Olszewski, et al. (2007). "TNF Trafficking to Human Mast Cell Granules: Mature Chain-Dependent Endocytosis". The Journal of Immunology. 178 (9): 5701–5709. doi:10.4049/jimmunol.178.9.5701. PMID 17442953.
- Walsh LJ, Trinchieri G, Waldorf HA, Whitaker D, Murphy GF (May 1991). "Human dermal mast cells contain and release tumor necrosis factor alpha, which induces endothelial leukocyte adhesion molecule 1". Proc. Natl. Acad. Sci. U.S.A. 88 (10): 4220–4. Bibcode:1991PNAS...88.4220W. doi:10.1073/pnas.88.10.4220. PMC 51630. PMID 1709737.
- Feng P, Jyotaki M, Kim A, Chai J, Simon N, Zhou M, Bachmanov AA, Huang L, Wang H (October 2015). "Regulation of bitter taste responses by tumor necrosis factor". Brain, Behavior, and Immunity. 49: 32–42. doi:10.1016/j.bbi.2015.04.001. PMC 4567432. PMID 25911043.
- Said EA, Dupuy FP, Trautmann L, Zhang Y, Shi Y, El-Far M, Hill BJ, Noto A, Ancuta P, Peretz Y, Fonseca SG, Van Grevenynghe J, Boulassel MR, Bruneau J, Shoukry NH, Routy JP, Douek DC, Haddad EK, Sekaly RP (April 2010). "Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection". Nat. Med. 16 (4): 452–9. doi:10.1038/nm.2106. PMC 4229134. PMID 20208540.
- Starkie R, Ostrowski SR, Jauffred S, Febbraio M, Pedersen BK (2003). "Exercise and IL-6 infusion inhibit endotoxin-induced TNF-α production in humans". FASEB J. 17 (8): 884–886. doi:10.1096/fj.02-0670fje. PMID 12626436.
- Pedersen BK (December 2009). "The diseasome of physical inactivity – and the role of myokines in muscle–fat cross talk". J Physiol. 587 (23): 5559–5568. doi:10.1113/jphysiol.2009.179515. PMC 2805368. PMID 19752112.
- Haraoui, B; Bykerk, V (2007). "Etanercept in the treatment of rheumatoid arthritis". Therapeutics and Clinical Risk Management. 3 (1): 99–105. doi:10.2147/tcrm.2007.3.1.99. PMC 1936291. PMID 18360618.
- Salomon BL, Leclerc M, Tosello J, Ronin E, Piaggio E, Cohen JL (2018). "Tumor Necrosis Factor α and Regulatory T Cells in Oncoimmunology". Front. Immunol. 9: 444. doi:10.3389/fimmu.2018.00444. PMC 5857565. PMID 29593717.
- Korneev, KV; Atretkhany, KN; Drutskaya, MS; Grivennikov, SI; Kuprash, DV; Nedospasov, SA (January 2017). "TLR-signaling and proinflammatory cytokines as drivers of tumorigenesis". Cytokine. 89: 127–135. doi:10.1016/j.cyto.2016.01.021. PMID 26854213.
- Bouwmeester T, Bauch A, Ruffner H, Angrand PO, Bergamini G, Croughton K, Cruciat C, Eberhard D, Gagneur J, Ghidelli S, Hopf C, Huhse B, Mangano R, Michon AM, Schirle M, Schlegl J, Schwab M, Stein MA, Bauer A, Casari G, Drewes G, Gavin AC, Jackson DB, Joberty G, Neubauer G, Rick J, Kuster B, Superti-Furga G (February 2004). "A physical and functional map of the human TNF alpha/NF-kappa B signal transduction pathway". Nat. Cell Biol. 6 (2): 97–105. doi:10.1038/ncb1086. PMID 14743216.
- Micheau O, Tschopp J (July 2003). "Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes". Cell. 114 (2): 181–90. doi:10.1016/S0092-8674(03)00521-X. PMID 12887920.
- Clark IA (June–August 2007). "How TNF was recognized as a key mechanism of disease". Cytokine Growth Factor Rev. 18 (3–4): 335–343. doi:10.1016/j.cytogfr.2007.04.002. PMID 17493863.
External links
- "Tasonermin". Drug Information Portal. U.S. National Library of Medicine.
- "Tumor Necrosis Factor-alpha". Drug Information Portal. U.S. National Library of Medicine.
- Tumor Necrosis Factor-alpha at the US National Library of Medicine Medical Subject Headings (MeSH)
- Overview of all the structural information available in the PDB for UniProt: P01375 (Tumor necrosis factor) at the PDBe-KB.
PDB gallery | |
|---|---|
|