SNARE (protein)
SNARE proteins — "SNAP REceptor" — are a large protein family consisting of at least 24 members in yeasts and more than 60 members in mammalian cells.[2][3] The primary role of SNARE proteins is to mediate vesicle fusion – the fusion of vesicles with the target membrane; this notably mediates exocytosis, but can also mediate the fusion of vesicles with membrane-bound compartments (such as a lysosome). The best studied SNAREs are those that mediate the neurotransmitter release of synaptic vesicles in neurons. These neuronal SNAREs are the targets of the neurotoxins responsible for botulism and tetanus produced by certain bacteria.

| SNARE-fusion membrane complex proteins | |
|---|---|
| Identifiers | |
| Symbol | SNARE |
| InterPro | IPR010989 |
| SCOPe | 1kil / SUPFAM |
| TCDB | 1.F.1 |
| OPM superfamily | 197 |
| OPM protein | 3hd7 |
| Membranome | 198 |
Types
SNAREs can be divided into two categories: vesicle or v-SNAREs, which are incorporated into the membranes of transport vesicles during budding, and target or t-SNAREs, which are associated with nerve terminal membranes. Evidence suggests that t-SNAREs form stable subcomplexes which serve as guides for v-SNARE, incorporated into the membrane of a protein-coated vesicle, binding to complete the formation of the SNARE complex.[4] Several SNARE proteins are located on both vesicles and target membranes, therefore, a more recent classification scheme takes into account structural features of SNAREs, dividing them into R-SNAREs and Q-SNAREs. Often, R-SNAREs act as v-SNAREs and Q-SNAREs act as t-SNAREs. R-SNAREs are proteins that contribute an arginine (R) residue in the formation of the zero ionic layer in the assembled core SNARE complex. One particular R-SNARE is synaptobrevin, which is located in the synaptic vesicles. Q-SNAREs are proteins that contribute a glutamine (Q) residue in the formation of the zero ionic layer in the assembled core SNARE complex. Q-SNAREs include syntaxin and SNAP-25. Q-SNAREs are further classified as Qa, Qb, or Qc depending on their location in the four-helix bundle.
Structure
SNAREs are small, abundant, sometimes tail-anchored proteins which are often post-translationally inserted into membranes via a C-terminal transmembrane domain. Seven of the 38 known SNAREs, including SNAP-25, do not have a transmembrane domain and are instead attached to the membrane via lipid modifications such as palmitoylation.[5] Tail-anchored proteins can be inserted into the plasma membrane, endoplasmic reticulum, mitochondria, and peroxisomes among other membranes, though any particular SNARE is targeted to a unique membrane. The targeting of SNAREs is accomplished by altering either the composition of the C-terminal flanking amino acid residues or the length of the transmembrane domain. Replacement of the transmembrane domain with lipid anchors leads to an intermediate stage of membrane fusion where only the two contacting leaflets fuse and not the two distal leaflets of the two membrane bilayer.[6]
Although SNAREs vary considerably in structure and size, they all share a segment in their cytosolic domain called a SNARE motif that consists of 60-70 amino acids and contains heptad repeats that have the ability to form coiled-coil structures. V- and t-SNAREs are capable of reversible assembly into tight, four-helix bundles called "trans"-SNARE complexes. In synaptic vesicles, the readily-formed metastable "trans" complexes are composed of three SNAREs: syntaxin 1 and SNAP-25 resident in cell membrane and synaptobrevin (also referred to as vesicle-associated membrane protein or VAMP) anchored in the vesicle membrane.
In neuronal exocytosis, syntaxin and synaptobrevin are anchored in respective membranes by their C-terminal domains, whereas SNAP-25 is tethered to the plasma membrane via several cysteine-linked palmitoyl chains. The core trans-SNARE complex is a four--helix bundle, where one -helix is contributed by syntaxin 1, one -helix by synaptobrevin and two -helices are contributed by SNAP-25.

The plasma membrane-resident SNAREs have been shown to be present in distinct microdomains or clusters, the integrity of which is essential for the exocytotic competence of the cell.
Membrane fusion
During membrane fusion, v-SNARE and t-SNARE proteins on separate membranes combine to form a trans-SNARE complex, also known as a "SNAREpin". Depending on the stage of fusion of the membranes, these complexes may be referred to differently.
During fusion of trans-SNARE complexes, the membranes merge and SNARE proteins involved in complex formation after fusion are then referred to as a "cis"-SNARE complex, because they now reside in a single (or cis) resultant membrane. After fusion, the cis-SNARE complex is bound and disassembled by an adaptor protein, alpha-SNAP. Then, the hexameric ATPase (of the AAA type) called NSF catalyzes the ATP-dependent unfolding of the SNARE proteins and releases them into the cytosol for recycling.
SNAREs are thought to be the core required components of the fusion machinery and can function independently of additional cytosolic accessory proteins. This was demonstrated by engineering "flipped" SNAREs, where the SNARE domains face the extracellular space rather than the cytosol. When cells containing v-SNAREs contact cells containing t-SNAREs, trans-SNARE complexes form and cell-cell fusion ensues.[7]
Components
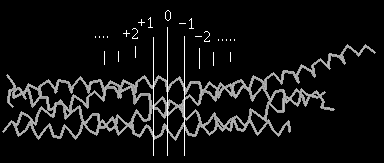
The core SNARE complex is a 4--helix bundle.[8] Synaptobrevin and syntaxin contribute one -helix each, while SNAP-25 participates with two -helices (abbreviated as Sn1 and Sn2). The interacting amino acid residues that zip the SNARE complex can be grouped into layers. Each layer has 4 amino acid residues - one residue per each of the 4 -helices. In the center of the complex is the zero ionic layer composed of one arginine (R) and three glutamine (Q) residues, and it is flanked by leucine zippering. Layers '-1', '+1' and '+2' at the centre of the complex most closely follow ideal leucine-zipper geometry and aminoacid composition.[9]
The zero ionic layer is composed of R56 from VAMP-2, Q226 from syntaxin-1A, Q53 from Sn1 and Q174 from Sn2, and is completely buried within the leucine-zipper layers. The positively charged guanidino group of the arginine (R) residue interact with the carboxyl groups of each of the three glutamine (Q) residues.
The flanking leucine-zipper layers act as a water-tight seal to shield the ionic interactions from the surrounding solvent. Exposure of the zero ionic layer to the water solvent by breaking the flanking leucine zipper leads to instability of the SNARE complex and is the putative mechanism by which -SNAP and NSF recycle the SNARE complexes after the completion of synaptic vesicle exocytosis.
Mechanism of membrane fusion
Assembly
SNARE proteins must assemble into trans-SNARE complexes to provide the force that is necessary for vesicle fusion. The four α-helix domains (1 each from synaptobrevin and syntaxin, and 2 from SNAP-25) come together to form a coiled-coil motif. The rate-limiting step in the assembly process is the association of the syntaxin SNARE domain, since it is usually found in a "closed" state where it is incapable of interacting with other SNARE proteins.[10] When syntaxin is in an open state, trans-SNARE complex formation begins with the association of the four SNARE domains at their N-termini. The SNARE domains proceed in forming a coiled-coil motif in the direction of the C-termini of their respective domains.
The SM protein Munc18 is thought to play a role in assembly of the SNARE complex, although the exact mechanism by which it acts is still under debate. It is known that the clasp of Munc18 locks syntaxin in a closed conformation by binding to its α-helical SNARE domains, which inhibits syntaxin from entering SNARE complexes (thereby inhibiting fusion).[10] The clasp is also capable, however, of binding the entire four-helix bundle of the trans-SNARE complex. One hypothesis suggests that, during SNARE-complex assembly, the Munc18 clasp releases closed syntaxin, remains associated with the N-terminal peptide of syntaxin (allowing association of the syntaxin SNARE domain with other SNARE proteins), and then reattaches to the newly formed four-helix SNARE complex.[11] This possible mechanism of dissociation and subsequent re-association with the SNARE domains could be calcium-dependent.[12] This supports the idea that Munc18 plays a key regulatory role in vesicle fusion; under normal conditions the SNARE complex will be prevented from forming by Munc18, but when triggered the Munc18 will actually assist in SNARE-complex assembly and thereby act as a fusion catalyst.[11]
Zippering and fusion pore opening
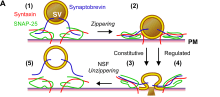
Membrane fusion is an energetically demanding series of events, which requires translocation of proteins in the membrane and disruption of the lipid bilayer, followed by reformation of a highly curved membrane structure. The process of bringing together two membranes requires input energy to overcome the repulsive electrostatic forces between the membranes. The mechanism that regulates the movement of membrane associated proteins away from the membrane contact zone prior to fusion is unknown, but the local increase in membrane curvature is thought to contribute in the process. SNAREs generate energy through protein-lipid and protein-protein interactions which act as a driving force for membrane fusion.
One model hypothesizes that the force required to bring two membranes together during fusion comes from the conformational change in trans-SNARE complexes to form cis-SNARE complexes. The current hypothesis that describes this process is referred to as SNARE "zippering."[13]
When the trans-SNARE complex is formed, the SNARE proteins are still found on opposing membranes. As the SNARE domains continue coiling in a spontaneous process, they form a much tighter, more stable four-helix bundle. During this "zippering" of the SNARE complex, a fraction of the released energy from binding is thought to be stored as molecular bending stress in the individual SNARE motifs. This mechanical stress is postulated to be stored in the semi-rigid linker regions between the transmembrane domains and the SNARE helical bundle.[14][15] The energetically unfavorable bending is minimized when the complex moves peripherally to the site of membrane fusion. As a result, relief of the stress overcomes the repulsive forces between the vesicle and the cell membrane and presses the two membranes together.[16]
Several models to explain the subsequent step - the formation of stalk and fusion pore - have been proposed. However, the exact nature of these processes remains debated. In accordance with the "zipper" hypothesis, as the SNARE complex forms, the tightening helix bundle puts torsional force on the transmembrane (TM) domains domains of synaptobrevin and syntaxin.[17] This causes the TM domains to tilt within the separate membranes as the proteins coil more tightly. The unstable configuration of the TM domains eventually causes the two membranes to fuse and the SNARE proteins come together within the same membrane, which is referred to as a "cis"-SNARE complex.[18] As a result of the lipid rearrangement, a fusion pore opens and allows the chemical contents of the vesicle to leak into the outside environment.
The continuum explanation of stalk formation suggests that membrane fusion begins with an infinitesimal radius until it radially expands into a stalk-like structure. However, such a description fails to take into account the molecular dynamics of membrane lipids. Recent molecular simulations show that the close proximity of the membranes allows the lipids to splay, where a population of lipids insert their hydrophobic tails into the neighboring membrane - effectively keeping a "foot" in each membrane. The resolution of the splayed lipid state proceeds spontaneously to form the stalk structure. In this molecular view, the splayed-lipid intermediate state is the rate determining barrier rather than the formation of the stalk, which now becomes the free energy minimum. The energetic barrier for establishment of the splayed-lipid conformation is directly proportional to the intermembrane distance. The SNARE complexes and their pressing of the two membranes together, therefore, could provide the free energy required to overcome the barrier.[19]
Disassembly
The energy input that is required for SNARE-mediated fusion to take place comes from SNARE-complex disassembly. The suspected energy source is N-ethylmaleimide-sensitive factor (NSF), an ATPase that is involved with membrane fusion. NSF homohexamers, along with the NSF cofactor α-SNAP, bind and dissociate the SNARE complex by coupling the process with ATP hydrolysis.[20] This process allows for reuptake of synaptobrevin for further use in vesicles, whereas the other SNARE proteins remain associated with the cell membrane.
The dissociated SNARE proteins have a higher energy state than the more stable cis-SNARE complex. It is believed that the energy that drives fusion is derived from the transition to a lower energy cis-SNARE complex. The ATP hydrolysis-coupled dissociation of SNARE complexes is an energy investment that can be compared to "cocking the gun" so that, once vesicle fusion is triggered, the process takes place spontaneously and at optimum velocity. A comparable process takes place in muscles, in which the myosin heads must first hydrolyze ATP in order to adapt the necessary conformation for interaction with actin and the subsequent power stroke to occur.
Regulatory effects on exocytosis
Regulation via SNAP-25 palmitoylation
The Q-SNARE protein Synaptosomal-associated protein 25 (SNAP-25) is composed of two α-helical domains connected by a random coil linker. The random coil linker region is most notable for its four cysteine residues.[21] The α-helical domains combine with those of both syntaxin and synaptobrevin (also known as vesicle associated membrane protein or VAMP) to form the 4-α-helix coiled-coil SNARE complex critical to efficient exocytosis.
While syntaxin and synaptobrevin both contain transmembrane domains which allow for docking with target and vesicle membranes respectively, SNAP-25 relies on the palmitoylation of cysteine residues found in its random coil region for docking to the target membrane. Some studies have suggested that association with syntaxin via SNARE interactions precludes the need for such docking mechanisms. Syntaxin knockdown studies however, failed to show a decrease in membrane bound SNAP-25 suggesting alternate docking means exist.[22] The covalent bonding of fatty acid chains to SNAP-25 via thioester linkages with one or more cysteine residues therefore, provides for regulation of docking and ultimately SNARE mediated exocytosis. This process is mediated by a specialized enzyme called DHHC palmitoyl transferase.[23] The cysteine rich domain of SNAP-25 has also been shown to weakly associate with the plasma membrane possibly allowing it to be localized near the enzyme for subsequent palmitoylation. The reverse of this process is carried out by another enzyme called palmitoyl protein thioesterase (see figure).
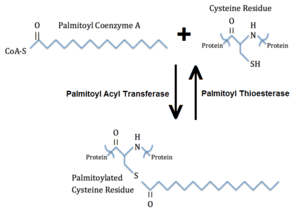
The availability of SNAP-25 in the SNARE complex is also theorized to possibly be spatially regulated via localization of lipid microdomains in the target membrane. Palmitoylated cysteine residues could be localized to the desired target membrane region via a favorable lipid environment (possibly cholesterol rich) complementary to the fatty acid chains bonded to the cysteine residues of SNAP-25.[24]
SNAP-25 regulation of voltage-gated Ca2+ channels in neuronal axon terminals
As an action potential reaches the axon terminal, depolarization events stimulate the opening of voltage-gated calcium channels (VGCCs) allowing the rapid influx of calcium down its electrochemical gradient. Calcium goes on to stimulate exocytosis via binding with synaptotagmin 1. SNAP-25 however, has been shown to negatively regulate VGCC function in glutamatergic neuronal cells. SNAP-25 leads to a reduction of current density through VGCC's and therefore a decrease in the amount of calcium that is binding the synaptotagmin, causing a decrease in neuronal glutamatergic exocytosis. Conversely, underexpression of SNAP-25 allows for an increase in VGCC current density and increase in exocytosis.[25]
Further investigation has suggested possible relationships between SNAP-25 over/underexpression and a variety of brain diseases. In attention-deficit/hyperactivity disorder or ADHD, polymorphisms at the SNAP-25 gene locus in humans have been linked to the disease suggesting a potential role in its manifestation.[26] This is further suggested by heterogeneous SNAP-25 knockout studies performed on coloboma mutant mice, which led to phenotypic characteristics of ADHD.[27] Studies have also shown a correlation of SNAP-25 over/underexpression and the onset of schizophrenia.[28][29]
Syntaxin and the Habc domain
Syntaxin consists of a transmembrane domain (TMD), alpha-helical SNARE domain, a short linker region, and the Habc domain which consists of three alpha-helical regions. The SNARE domain in syntaxin serves as a target site for docking of SNAP-25 and synaptobrevin in order to form the four helix bundle requisite to the SNARE complex and subsequent fusion. The Habc domain, however, serves as an autoinhibitory domain in syntaxin. It has been shown to fold over and associate with the SNARE domain of syntaxin inducing a "closed" state, creating a physical barrier to the formation of the SNARE motif. Conversely, the Habc domain can again disassociate with the SNARE domain leaving syntaxin free to associate with both SNAP-25 and synaptobrevin.[30]
Syntaxin 1B and readily releasable pool of vesicles
There is an immense diversity of syntaxin subtypes, with 15 varieties in the human genome.[31] It has been suggested that syntaxin1B has a role in regulating number of synaptic vesicles ready for exocytosis in the axon terminal. This is also called the readily releasable pool (RRP) of vesicles. A knock out study in 2014 showed that the lack of syntaxin1B led to a significant decrease in RRP size.[32]
Toxins
Many neurotoxins directly affect SNARE complexes. Such toxins as the botulinum and tetanus toxins work by targeting the SNARE components. These toxins prevent proper vesicle recycling and result in poor muscle control, spasms, paralysis, and even death.
Botulinum neurotoxin
Botulinum Toxin (BoNT) is one of the most potent toxins to have ever been discovered.[33] It is a proteolytic enzyme that cleaves SNARE proteins in neurons. Its protein structure is composed of two peptide subunits, a heavy chain (100kDas) and a light chain (50kDas), which are held together by a disulfide bond. The action of BoNT follows a 4-step mechanism including binding to the neuronal membrane, endocytosis, membrane translocation, and proteolysis of SNARE proteins.[34]
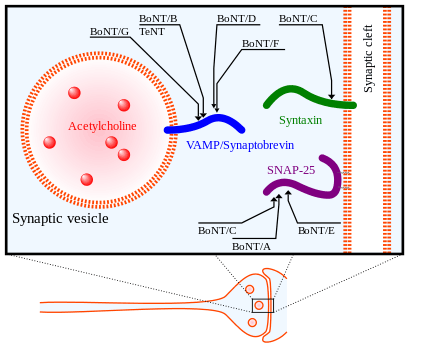
In its mechanism of action, the heavy chain of BoNT is first used to find its neuronal targets and bind to the gangliosides and membrane proteins of presynaptic neurons. Next, the toxin is then endocytosed into the cell membrane. The heavy chain undergoes a conformational change important for translocating the light chain into the cytosol of the neuron. Finally, after the light chain of BoNT is brought into the cytosol of the targeted neuron, it is released from the heavy chain so that it can reach its active cleavage sites on the SNARE proteins.[34] The light chain is released from the heavy chain by the reduction of the disulfide bond holding the two together. The reduction of this disulfide bond is mediated by the NADPH-thioredoxin reductase-thioredoxin system.[36] The light chain of BoNT acts as a metalloprotease on SNARE proteins that is dependent on Zn(II) ions,[37] cleaving them and eliminating their function in exocytosis.
There are 8 known isotypes of BoNT, BoNT/A - BoNT/H, each with different specific cleavage sites on SNARE proteins. SNAP25, a member of the SNARE protein family located in the membrane of cells, is cleaved by BoNT isotypes A, C, and E. The cleavage of SNAP-25 by these isotypes of BoNT greatly inhibits their function in forming the SNARE complex for fusion of vesicles to the synaptic membrane. BoNT/C also targets Syntaxin-1, another SNARE protein located in the synaptic membrane. It degenerates these Syntaxin proteins with a similar outcome as with SNAP-25. A third SNARE protein, Synaptobrevin (VAMP), is located on cell vesicles. VAMP2 is targeted and cleaved by BoNT isotypes B, D, and F in synaptic neurons.[33] The targets of these various isotypes of BoNT as well as Tetanus Neurotoxin (TeNT) are shown in the figure to the right.
In each of these cases, Botulinum Neurotoxin causes functional damage to SNARE proteins, which has significant physiological and medical implications. By damaging SNARE proteins, the toxin prevents synaptic vesicles from fusing to the synaptic membrane and releasing their neurotransmitters into the synaptic cleft. With the inhibition of neurotransmitter release into the synaptic cleft, action potentials cannot be propagated to stimulate muscle cells. This result in paralysis of those infected and in serious cases, it can cause death. Although the effects of Botulinum Neurotoxin can be fatal, it has also been used as a therapeutic agent in medical and cosmetic treatments.[38][39]
Tetanus neurotoxin
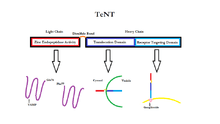
Tetanus toxin, or TeNT, is composed of a heavy chain (100KDa) and a light chain (50kDa) connected by a disulfide bond. The heavy chain is responsible for neurospecific binding of TeNT to the nerve terminal membrane, endocytosis of the toxin, and translocation of the light chain into the cytosol. The light chain has zinc-dependent endopeptidase or more specifically matrix metalloproteinase (MMP) activity through which cleaveage of synaptobrevin or VAMP is carried out.[40]
For the light chain of TeNT to be activated one atom of zinc must be bound to every molecule of toxin.[41] When zinc is bound reduction of the disulfide bond will be carried out primarily via the NADPH-thioredoxin reductase-thioredoxin redox system.[42] Then the light chain is free to cleave the Gln76-Phe77 bond of synaptobrevin.[40] Cleavage of synaptobrevin affects the stability of the SNARE core by restricting it from entering the low energy conformation which is the target for NSF binding.[43] This cleavage of synaptobrevin is the final target of TeNT and even in low doses the neurotoxin will inhibit neurotransmitter exocytosis.
Role in neurotransmitter release
Neurotransmitters are stored in readily releasable pools of vesicles confined within the presynaptic terminal. During neurosecretion/exocytosis, SNAREs play a crucial role in vesicle docking, priming, fusion, and synchronization of neurotransmitter release into the synaptic cleft.
The first step in synaptic vesicle fusion is tethering, where the vesicles are translocated from the reserve pool into physical contact with the membrane. At the membrane, Munc-18 is initially bound to syntaxin 1A in a closed structure. It is postulated that the dissociation of Munc-18 from the complex frees syntaxin 1A to bind with the v-SNARE proteins.[44] The next step in release is the docking of vesicles, where the v- and t-SNARE proteins transiently associate in a calcium-independent manner. The vesicles are then primed, wherein the SNARE motifs form a stable interaction between the vesicle and membrane. Complexins stabilize the primed SNARE-complex rendering the vesicles ready for rapid exocytosis.
The span of presynaptic membrane containing the primed vesicles and dense collection of SNARE proteins is referred to as the active zone. Voltage-gated calcium channels are highly concentrated around active zones and open in response to membrane depolarization at the synapse. The influx of calcium is sensed by synaptotagmin 1, which in turn dislodges complexin protein and allows the vesicle to fuse with the presynaptic membrane to release neurotransmitter. It has also been shown that the voltage-gated calcium channels directly interact with the t-SNAREs syntaxin 1A and SNAP-25, as well as with synaptotagmin 1. The interactions are able to inhibit calcium channel activity as well as tightly aggregate the molecules around the release site.[45]
There have been many clinical cases that link SNARE genes with neural disorders. Deficiency in SNAP-25 mRNA has been observed in hippocampal tissue of some schizophrenic patients, a SNAP-25 single-nucleotide polymorphism is linked to hyperactivity in autism-spectrum disorders, and overexpression of SNAP-25B leads to the early onset of bipolar disorder.[45]
Role in autophagy
Macroautophagy is a catabolic process involving the formation of double-membrane bound organelles called autophagosomes, which aid in degradation of cellular components through fusion with lysosomes. During autophagy, portions of the cytoplasm are engulfed by a cup-shaped double-membrane structure called a phagophore and eventually become the contents of the fully assembled autophagosome. Autophagosome biogenesis requires the initiation and growth of phagophores, a process that was once thought to occur through de novo addition of lipids. However, recent evidence suggests that the lipids that contribute to the growing phagophores originate from numerous sources of membrane, including endoplasmic reticulum, Golgi, plasma membrane, and mitochondria.[46] SNAREs play important roles in mediating vesicle fusion during phagophore initiation and expansion as well as autophagosome-lysosome fusion in the later stages of autophagy.
Though the mechanism of phagophore initiation in mammals is unknown, SNAREs have been implicated in phagophore formation through homotypic fusion of small, clathrin-coated, single-membrane vesicles containing Atg16L, the v-SNARE VAMP7, and its partner t-SNAREs: Syntaxin-7, Syntaxin-8, and VTI1B.[47] In yeast, the t-SNAREs Sec9p and Sso2p are required for exocytosis and promote tubulovesicular budding of Atg9 positive vesicles, which are also required for autophagosome biogenesis.[48][49] Knocking out either of these SNAREs leads to accumulation of small Atg9 containing vesicles that do not fuse, therefore preventing the formation of the pre-autophagosomal structure.[49]
In addition to phagophore assembly, SNAREs are also important in mediating autophagosome-lysosome fusion. In mammals, the SNAREs VAMP7, VAMP8, and VTI1B are required in autophagosome-lysosome fusion and this process is impaired in lysosomal storage disorders where cholesterol accumulates in the lysosome and sequesters SNAREs in cholesterol rich regions of the membrane preventing their recycling.[50] Recently, syntaxin 17 (STX17) was identified as an autophagosome associated SNARE that interacts with VAMP8 and SNAP29 and is required for fusion with the lysosome.[51] STX17 is localized on the outer membrane of autophagosomes, but not phagophores or other autophagosome precursors, which prevents them from prematurely fusing with the lysosome.[51] In yeast, the fusion of autophagosomes with vacuoles (the yeast equivalent of lysosomes) requires SNAREs and related proteins such as the syntaxin homolog Vam3, SNAP-25 homolog Vam7, Ras-like GTPase Ypt7, and the NSF ortholog, Sec18.[46]
References
- Georgiev, Danko D; James F . Glazebrook (2007). "Subneuronal processing of information by solitary waves and stochastic processes". In Lyshevski, Sergey Edward (ed.). Nano and Molecular Electronics Handbook. Nano and Microengineering Series. CRC Press. pp. 17–1–17–41. doi:10.1201/9781420008142.ch17 (inactive 2020-01-22). ISBN 978-0-8493-8528-5.
- Burri, Lena; Lithgow, Trevor (2004-01-01). "A complete set of SNAREs in yeast". Traffic. 5 (1): 45–52. doi:10.1046/j.1600-0854.2003.00151.x. ISSN 1398-9219. PMID 14675424.
- Gerald K (2002). Cell and Molecular Biology (4th ed.). John Wiley & Sons.
- Malsam J, Söllner TH (1 October 2011). "Organization of SNAREs within the Golgi stack". Cold Spring Harbor Perspectives in Biology. 3 (10): a005249. doi:10.1101/cshperspect.a005249. PMC 3179334. PMID 21768609.
- Hong W, Lev S (January 2014). "Tethering the assembly of SNARE complexes". Trends in Cell Biology. 24 (1): 35–43. doi:10.1016/j.tcb.2013.09.006. PMID 24119662.
- Martens S, McMahon HT (21 May 2008). "Mechanisms of membrane fusion: disparate players and common principles". Nature Reviews Molecular Cell Biology. 9 (7): 543–556. doi:10.1038/nrm2417. PMID 18496517.
- Hu C, Ahmed M, Melia TJ, Söllner TH, Mayer T, Rothman JE (13 June 2003). "Fusion of Cells by Flipped SNAREs". Science. 300 (5626): 1745–1749. doi:10.1126/science.1084909. PMID 12805548.
- Sutton RB, Fasshauer D, Jahn R, Brunger AT (1998). "Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution". Nature. 395 (6700): 347–353. doi:10.1038/26412. PMID 9759724.
- Fasshauer D, Sutton RB, Brunger AT, Jahn R (1998). "Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs". Proceedings of the National Academy of Sciences. 95 (26): 15781–15786. doi:10.1073/pnas.95.26.15781. PMC 28121. PMID 9861047.
- Burkhardt P, Hattendorf DA, Weis WI, Fasshauer D (2008). "Munc18a controls SNARE assembly through its interaction with the syntaxin N-peptide". EMBO J. 27 (7): 923–33. doi:10.1038/emboj.2008.37. PMC 2323264. PMID 18337752.
- Südhof TC, Rothman JE (January 2009). "Membrane fusion: grappling with SNARE and SM proteins". Science. 323 (5913): 474–7. doi:10.1126/science.1161748. PMC 3736821. PMID 19164740.
- Jahn R, Fasshauer D (2012). "Molecular machines governing exocytosis of synaptic vesicles". Nature. 490 (7419): 201–7. doi:10.1038/nature11320. PMC 4461657. PMID 23060190.
- Chen YA, Scheller RH (2001). "SNARE-mediated membrane fusion". Nat. Rev. Mol. Cell Biol. 2 (2): 98–106. doi:10.1038/35052017. PMID 11252968.
- Wang Y, Dulubova I, Rizo J, Südhof TC (2001). "Functional analysis of conserved structural elements in yeast syntaxin Vam3p". J. Biol. Chem. 276 (30): 28598–605. doi:10.1074/jbc.M101644200. PMID 11349128.
- Kiessling V, Tamm LK (January 2003). "Measuring distances in supported bilayers by fluorescence interference-contrast microscopy: polymer supports and SNARE proteins". Biophysical Journal. 84 (1): 408–18. doi:10.1016/s0006-3495(03)74861-9. PMC 1302622. PMID 12524294.
- Risselada HJ, Kutzner C, Grubmüller H (2 May 2011). "Caught in the act: visualization of SNARE-mediated fusion events in molecular detail". ChemBioChem: A European Journal of Chemical Biology. 12 (7): 1049–55. doi:10.1002/cbic.201100020. hdl:11858/00-001M-0000-0027-C8EA-9. PMID 21433241.
- Fang Q, Lindau M (2014). "How could SNARE proteins open a fusion pore?". Physiology. 29 (4): 278–85. doi:10.1152/physiol.00026.2013. PMC 4103061. PMID 24985331.
- Zucker, Robert S.; Kullmann, Dimitri M.; Kaeser, Pascal S. (August 2014). "Chapter 15: Release of Neurotransmitters". In Byrne, John H.; Heidelberger, Ruth; Waxham, M. Neal (eds.). From Molecules to Networks: An Introduction to Cellular and Molecular Neuroscience. Academic Press. pp. 443–488. ISBN 9780123982674. External link in
|title=(help) - Risselada HJ, Grubmüller H (April 2012). "How SNARE molecules mediate membrane fusion: recent insights from molecular simulations". Current Opinion in Structural Biology. 22 (2): 187–96. doi:10.1016/j.sbi.2012.01.007. hdl:11858/00-001M-0000-000F-9AF7-9. PMID 22365575.
- Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE (1993). "A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion". Cell. 75 (3): 409–18. doi:10.1016/0092-8674(93)90376-2. PMID 8221884.
- Bock, LV; Woodbury, DJ (9 August 2010). "Chemomechanical regulation of SNARE proteins studied with molecular dynamics simulations". Biophysical Journal. 99 (4): 1221–1230. doi:10.1016/j.bpj.2010.06.019. PMC 2920728. PMID 20713006.
- Greaves, Jennifer (5 April 2009). "Regulation of SNAP-25 Trafficking and Function by Palmitoylation". Biochemical Society Transactions. 38 (part 1): 163–166. doi:10.1042/BST0380163. PMID 20074052.
- Greaves, Jennifer (11 May 2010). "Palmitoylation of the SNAP-25 Protein Family: Specificity and Regulation by DHHC Palmitoyl Transferases". The Journal of Biological Chemistry. 285 (32): 24629–24638. doi:10.1074/jbc.M110.119289. PMC 2915699. PMID 20519516.
- Greaves, Jennifer (5 April 2009). "Regulation of SNAP-25 Trafficking and Function by Palmitoylation". Biochemical Society Transactions. 38 (part 1): 163–166. doi:10.1042/bst0380163. PMID 20074052.
- Condliffe, Steven B (3 June 2010). "Endogenous SNAP-25 Regulates Native Voltage-gated Calcium Channels in Glutamatergic Neurons". The Journal of Biological Chemistry. 285 (32): 24968–24976. doi:10.1074/jbc.M110.145813. PMC 2915732. PMID 20522554.
- Corradini, Irene (21 January 2009). "SNAP-25 in Neuropsychiatric Disorders". Annals of the New York Academy of Sciences. 1152: 93–99. doi:10.1111/j.1749-6632.2008.03995.x. PMC 2706123. PMID 19161380.
- Hess, EJ (1992). "Spontaneous locomotor hyperactivity in a mouse mutant with a deletion including the Snap gene on chromosome 2". Journal of Neuroscience. 12 (7): 2865–2874. doi:10.1523/JNEUROSCI.12-07-02865.1992. PMC 6575838. PMID 1613559.
- Thompson, PM (1998). "Altered levels of the synaptosomal associated protein SNAP-25 in schizophrenia". Biological Psychiatry. 43 (4): 239–243. doi:10.1016/s0006-3223(97)00204-7. PMID 9513732.
- Gabriel, SM (1997). "Increased concentrations of presynaptic proteins in the cingulate cortex of subjects with schizophrenia". Archives of General Psychiatry. 54 (6): 559–566. doi:10.1001/archpsyc.1997.01830180077010. PMID 9193197.
- MacDonald, Chris (3 April 2009). "Autoinhibition of SNARE complex assembly by a conformational switch represents a conserved feature of syntaxins". Biochemical Society Transactions. 38 (Pt 1): 209–212. doi:10.1042/BST0380209. PMC 5242387. PMID 20074061.
- Teng, Felicia Yu Hsuan (24 October 2001). "The Syntaxin". Genome Biology. 2 (11): reviews 3012.1–7. doi:10.1186/gb-2001-2-11-reviews3012. PMC 138984. PMID 11737951.
- Mishima, Tatsuya (28 February 2014). "Syntaxin 1B, but Not Syntaxin 1A, Is Necessary for the Regulation of Synaptic Vesicle Exocytosis and of the Readily Releasable Pool at Central Synapses". PLoS ONE. 9 (2): e90004. doi:10.1371/journal.pone.0090004. PMC 3938564. PMID 24587181.
- Peng L, Liu H, Ruan H, Tepp WH, Stoothoff WH, Brown RH, Johnson EA, Yao WD, Zhang SC, Dong M (12 February 2013). "Cytotoxicity of botulinum neurotoxins reveals a direct role of syntaxin 1 and SNAP-25 in neuron survival". Nature Communications. 4: 1472. doi:10.1038/ncomms2462. PMC 4052923. PMID 23403573.
- Rossetto O, Pirazzini M, Bolognese P, Rigoni M, Montecucco C (December 2011). "An update on the mechanism of action of tetanus and botulinum neurotoxins" (PDF). Acta Chim Slov. 58 (4): 702–7. PMID 24061118.
- Barr JR, Moura H, Boyer AE, Woolfitt AR, Kalb SR, Pavlopoulos A, McWilliams LG, Schmidt JG, Martinez RA, Ashley DL (2005). "Botulinum neurotoxin detection and differentiation by mass spectrometry". Emerging Infect. Dis. 11 (10): 1578–83. doi:10.3201/eid1110.041279. PMC 3366733. PMID 16318699.
- Pirazzini M, Bordin F, Rossetto O, Shone CC, Binz T, Montecucco C (January 2013). "The thioredoxin reductase-thioredoxin system is involved in the entry of tetanus and botulinum neurotoxins in the cytosol of nerve terminals". FEBS Letters. 587 (2): 150–155. doi:10.1016/j.febslet.2012.11.007. PMID 23178719.
- Silvaggi NR, Wilson D, Tzipori S, Allen KN (May 2008). "Catalytic Features of the Botulinum Neurotoxin A light chain Revealed by High Resolution Structure of an Inhibitory Peptide Complex". Biochemistry. 47 (21): 5736–5745. doi:10.1021/bi8001067. PMID 18457419.
- Wheeler AH (1998). "Botulinum toxin A, adjunctive therapy for refractory headaches associated with pericranial muscle tension". Headache. 38 (6): 468–71. doi:10.1046/j.1526-4610.1998.3806468.x. PMID 9664753.
- Garcia A, Fulton JE (1996). "Cosmetic denervation of the muscles of facial expression with botulinum toxin. A dose-response study". Dermatol Surg. 22 (1): 39–43. doi:10.1111/j.1524-4725.1996.tb00569.x. PMID 8556256.
- Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, Montecucco C (29 October 1992). "Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin". Nature. 359 (6398): 832–5. doi:10.1038/359832a0. PMID 1331807.
- Schiavo G, Poulain B, Rossetto O, Benfenati F, Tauc L, Montecucco C (October 1992). "Tetanus toxin is a zinc protein and its inhibition of neurotransmitter release and protease activity depend on zinc". The EMBO Journal. 11 (10): 3577–83. doi:10.1002/j.1460-2075.1992.tb05441.x. PMC 556816. PMID 1396558.
- Pirazzini M, Bordin F, Rossetto O, Shone CC, Binz T, Montecucco C (2013). "The thioredoxin reductase-thioredoxin system is involved in the entry of tetanus and botulinum neurotoxins in the cytosol of nerve terminals". FEBS Lett. 587 (2): 150–5. doi:10.1016/j.febslet.2012.11.007. PMID 23178719.
- Pellegrini LL, O'Connor V, Lottspeich F, Betz H (2 October 1995). "Clostridial neurotoxins compromise the stability of a low energy SNARE complex mediating NSF activation of synaptic vesicle fusion". The EMBO Journal. 14 (19): 4705–13. doi:10.1002/j.1460-2075.1995.tb00152.x. PMC 394567. PMID 7588600.
- Shi L, Kümmel D, Coleman J, Melia TJ, Giraudo CG (November 2011). "Dual roles of Munc18-1 rely on distinct binding modes of the central cavity with Stx1A and SNARE complex". Molecular Biology of the Cell. 22 (21): 4150–60. doi:10.1091/mbc.e11-02-0150. PMC 3204075. PMID 21900493.
- Ramakrishnan NA, Drescher MJ, Drescher DG (May 2012). "The SNARE complex in neuronal and sensory cells". Molecular and Cellular Neurosciences. 50 (1): 58–69. doi:10.1016/j.mcn.2012.03.009. PMC 3570063. PMID 22498053.
- Moreau K, Ravikumar B, Renna M, Puri C, Rubinsztein DC (July 2011). "Autophagosome Precursor Maturation Requires Homotypic Fusion". Cell. 146 (2): 303–317. doi:10.1016/j.cell.2011.06.023. PMC 3171170. PMID 21784250.
- Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC (18 July 2010). "Plasma membrane contributes to the formation of pre-autophagosomal structures". Nature Cell Biology. 12 (8): 747–757. doi:10.1038/ncb2078. PMC 2923063. PMID 20639872.
- Abeliovich, Hagai (1999). "Cytoplasm to vacuole trafficking of aminopeptidase I requires a t-SNARE/Sec1 complex composed of Tlg2 and Vps45". EMBO Journal. 18 (21): 6005–6016. doi:10.1093/emboj/18.21.6005. PMC 1171666. PMID 10545112.
- Nair U, Jotwani A, Geng J, Gammoh N, Richerson D, Yen WL, Griffith J, Nag S, Wang K, Moss T, Baba M, McNew JA, Jiang X, Reggiori F, Melia TJ, Klionsky DJ (July 2011). "SNARE Proteins Are Required for Macroautophagy". Cell. 146 (2): 290–302. doi:10.1016/j.cell.2011.06.022. PMC 3143362. PMID 21784249.
- Fraldi A, Annunziata F, Lombardi A, Kaiser HJ, Medina DL, Spampanato C, Fedele AO, Polishchuk R, Sorrentino NC, Simons K, Ballabio A (24 September 2010). "Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders". The EMBO Journal. 29 (21): 3607–3620. doi:10.1038/emboj.2010.237. PMC 2982760. PMID 20871593.
- Itakura E, Kishi-Itakura C, Mizushima N (December 2012). "The Hairpin-type Tail-Anchored SNARE Syntaxin 17 Targets to Autophagosomes for Fusion with Endosomes/Lysosomes". Cell. 151 (6): 1256–1269. doi:10.1016/j.cell.2012.11.001. PMID 23217709.
External links
- SNARE+Proteins at the US National Library of Medicine Medical Subject Headings (MeSH)
- SNARE+Complex at the US National Library of Medicine Medical Subject Headings (MeSH)