Sickle cell disease
Sickle cell disease (SCD) is a group of blood disorders typically inherited from a person's parents.[2] The most common type is known as sickle cell anaemia (SCA).[2] It results in an abnormality in the oxygen-carrying protein haemoglobin found in red blood cells.[2] This leads to a rigid, sickle-like shape under certain circumstances.[2] Problems in sickle cell disease typically begin around 5 to 6 months of age.[1] A number of health problems may develop, such as attacks of pain ("sickle cell crisis"), anemia, swelling in the hands and feet, bacterial infections and stroke.[1] Long-term pain may develop as people get older.[2] The average life expectancy in the developed world is 40 to 60 years.[2]
| Sickle cell disease | |
|---|---|
| Other names | Sickle cell disorder |
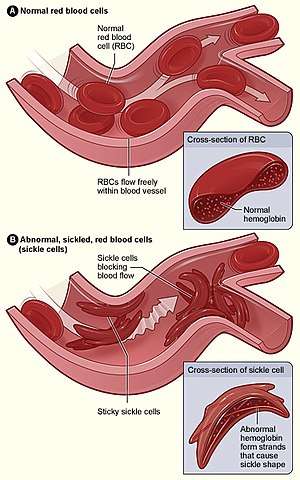 | |
| Figure (A) shows normal red blood cells flowing freely through veins. The inset shows a cross section of a normal red blood cell with normal haemoglobin. Figure (B) shows abnormal, sickled red blood cells sticking at the branching point in a vein. The inset image shows a cross-section of a sickle cell with long polymerized sickle haemoglobin (HbS) strands stretching and distorting the cell shape to look like a crescent. | |
| Specialty | Hematology |
| Symptoms | Attacks of pain, anemia, swelling in the hands and feet, bacterial infections, stroke[1] |
| Complications | Chronic pain, stroke, aseptic bone necrosis, gallstones, leg ulcers, priapism, pulmonary hypertension, vision problems, kidney problems[2] |
| Usual onset | 5–6 months of age[1] |
| Causes | Genetic[3] |
| Diagnostic method | Blood test[4] |
| Treatment | Vaccination, antibiotics, high fluid intake, folic acid supplementation, pain medication, blood transfusions[5][6] |
| Prognosis | Life expectancy 40–60 years (developed world)[2] |
| Frequency | 4.4 million (2015)[7] |
| Deaths | 114,800 (2015)[8] |
Sickle cell disease occurs when a person inherits two abnormal copies of the β-globin gene that makes haemoglobin, one from each parent.[3] This gene occurs in chromosome 11.[9] Several subtypes exist, depending on the exact mutation in each haemoglobin gene.[2] An attack can be set off by temperature changes, stress, dehydration, and high altitude.[1] A person with a single abnormal copy does not usually have symptoms and is said to have sickle cell trait.[3] Such people are also referred to as carriers.[5] Diagnosis is by a blood test, and some countries test all babies at birth for the disease.[4] Diagnosis is also possible during pregnancy.[4]
The care of people with sickle cell disease may include infection prevention with vaccination and antibiotics, high fluid intake, folic acid supplementation, and pain medication.[5][6] Other measures may include blood transfusion and the medication hydroxycarbamide (hydroxyurea).[6] A small percentage of people can be cured by a transplant of bone marrow cells.[2]
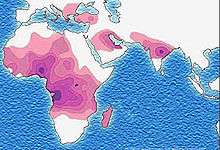
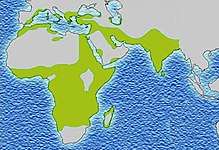
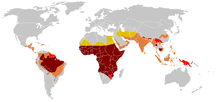
As of 2015, about 4.4 million people have sickle cell disease, while an additional 43 million have sickle cell trait.[7][10] About 80% of sickle cell disease cases are believed to occur in Sub-Saharan Africa.[11] It also occurs relatively frequently in parts of India, the Arabian Peninsula, and among people of African origin living in other parts of the world.[12] In 2015, it resulted in about 114,800 deaths.[8] The condition was first described in the medical literature by American physician James B. Herrick in 1910.[13][14] In 1949, its genetic transmission was determined by E. A. Beet and J. V. Neel.[14] In 1954, the protective effect against malaria of sickle cell trait was described.[14]
Signs and symptoms
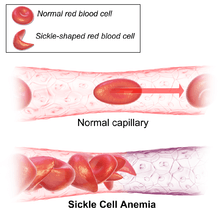
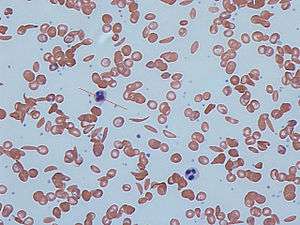
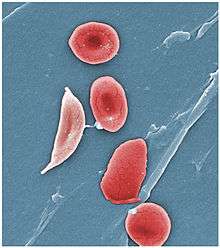
Signs of sickle cell disease usually begin in early childhood. The severity of symptoms can vary from person to person.[15] Sickle cell disease may lead to various acute and chronic complications, several of which have a high mortality rate.[16]
Sickle cell crisis
The terms "sickle cell crisis" or "sickling crisis" may be used to describe several independent acute conditions occurring in patients with SCD, which results in anaemia and crises that could be of many types, including the vaso-occlusive crisis, aplastic crisis, sequestration crisis, haemolytic crisis, and others. Most episodes of sickle cell crises last between five and seven days.[17] "Although infection, dehydration, and acidosis (all of which favor sickling) can act as triggers, in most instances, no predisposing cause is identified."[18]
Vaso-occlusive crisis
The vaso-occlusive crisis is caused by sickle-shaped red blood cells that obstruct capillaries and restrict blood flow to an organ, resulting in ischaemia, pain, necrosis, and often organ damage. The frequency, severity, and duration of these crises vary considerably. Painful crises are treated with hydration, analgesics, and blood transfusion; pain management requires opioid drug administration at regular intervals until the crisis has settled. For milder crises, a subgroup of patients manages on nonsteroidal anti-inflammatory drugs such as diclofenac or naproxen. For more severe crises, most patients require inpatient management for intravenous opioids; patient-controlled analgesia devices are commonly used in this setting. Vaso-occlusive crisis involving organs such as the penis[19] or lungs are considered an emergency and treated with red blood cell transfusions. Incentive spirometry, a technique to encourage deep breathing to minimise the development of atelectasis, is recommended.[20]
Splenic sequestration crisis
Because of its narrow vessels and function in clearing defective red blood cells, the spleen is frequently affected.[21] It is usually infarcted before the end of childhood in individuals suffering from sickle cell anaemia. This spleen damage increases the risk of infection from encapsulated organisms;[22][23] preventive antibiotics and vaccinations are recommended for those lacking proper spleen function.
Splenic sequestration crises are acute, painful enlargements of the spleen, caused by intrasplenic trapping of red cells and resulting in a precipitous fall in haemoglobin levels with the potential for hypovolemic shock. Sequestration crises are considered an emergency. If not treated, patients may die within 1–2 hours due to circulatory failure. Management is supportive, sometimes with blood transfusion. These crises are transient; they continue for 3–4 hours and may last for one day.[24]
Acute chest syndrome
Acute chest syndrome is defined by at least two of these signs or symptoms: chest pain, fever, pulmonary infiltrate or focal abnormality, respiratory symptoms, or hypoxemia.[25] It is the second-most common complication and it accounts for about 25% of deaths in patients with SCD. Most cases present with vaso-occlusive crises, and then develop acute chest syndrome.[26][27] Nevertheless, about 80% of people have vaso-occlusive crises during acute chest syndrome.
Aplastic crisis
Aplastic crises are acute worsenings of the patient's baseline anaemia, producing pale appearance, fast heart rate, and fatigue. This crisis is normally triggered by parvovirus B19, which directly affects production of red blood cells by invading the red cell precursors and multiplying in and destroying them.[28] Parvovirus infection almost completely prevents red blood cell production for two to three days. In normal individuals, this is of little consequence, but the shortened red cell life of SCD patients results in an abrupt, life-threatening situation. Reticulocyte counts drop dramatically during the disease (causing reticulocytopenia), and the rapid turnover of red cells leads to the drop in haemoglobin. This crisis takes 4 to 7 days to disappear. Most patients can be managed supportively; some need blood transfusion.[29]
Haemolytic crisis
Haemolytic crises are acute accelerated drops in haemoglobin level. The red blood cells break down at a faster rate. This is particularly common in people with coexistent G6PD deficiency.[30] Management is supportive, sometimes with blood transfusions.[20]
Other
One of the earliest clinical manifestations is dactylitis, presenting as early as six months of age, and may occur in children with sickle cell trait.[31] The crisis can last up to a month.[32] Given that pneumonia and sickling in the lung can both produce symptoms of acute chest syndrome, the patient is treated for both conditions.[33] It can be triggered by painful crisis, respiratory infection, bone-marrow embolisation, or possibly by atelectasis, opiate administration, or surgery. Hematopoietic ulcers may also occur.[34]
Genetics
Normally, humans have haemoglobin A, which consists of two alpha and two beta chains, haemoglobin A2, which consists of two alpha and two delta chains, and haemoglobin F, consisting of two alpha and two gamma chains in their bodies. Of these three types, haemoglobin F dominates until about 6 weeks of age. Afterwards, haemoglobin A dominates throughout life.[35] In people diagnosed with sickle cell disease, at least one of the β-globin subunits in haemoglobin A is replaced with what is known as haemoglobin S. In sickle cell anaemia, a common form of sickle cell disease, haemoglobin S replaces both β-globin subunits in the haemoglobin.[15]
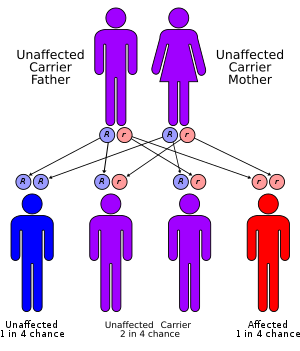
Sickle cell conditions have an autosomal recessive pattern of inheritance from parents.[36] The types of haemoglobin a person makes in the red blood cells depend on what haemoglobin genes are inherited from her or his parents. If one parent has sickle cell anaemia and the other has sickle cell trait, then the child has a 50% chance of having sickle cell disease and a 50% chance of having sickle cell trait. When both parents have sickle cell trait, a child has a 25% chance of sickle cell disease; 25% do not carry any sickle cell alleles, and 50% have the heterozygous condition.[37]
Sickle cell gene mutation probably arose spontaneously in different geographic areas, as suggested by restriction endonuclease analysis. These variants are known as Cameroon, Senegal, Benin, Bantu, and Saudi-Asian. Their clinical importance is because some are associated with higher HbF levels, e.g., Senegal and Saudi-Asian variants, and tend to have milder disease.[38]
The gene defect is a single nucleotide mutation (see single-nucleotide polymorphism – SNP) (GAG codon changing to GTG) of the β-globin gene, which results in glutamic acid (E/Glu) being substituted by valine (V/Val) at position 6 (E6V substitution).[39][note 1] Haemoglobin S with this mutation is referred to as HbS, as opposed to the normal adult HbA. This is normally a benign mutation, causing no apparent effects on the secondary, tertiary, or quaternary structures of haemoglobin in conditions of normal oxygen concentration. However, under low oxygen concentration, HbS polymerizes and forms fibrous precipitates because the deoxy form of haemoglobin exposes a hydrophobic patch on the protein between the E and F helices (Phe 85, Leu 88).[40]
In people heterozygous for HbS (carriers of sickling haemoglobin), the polymerisation problems are minor because the normal allele is able to produce half of the haemoglobin. In people homozygous for HbS, the presence of long-chain polymers of HbS distort the shape of the red blood cell from a smooth, doughnut-like shape to ragged and full of spikes, making it fragile and susceptible to breaking within capillaries. Carriers have symptoms only if they are deprived of oxygen (for example, while climbing a mountain) or while severely dehydrated.
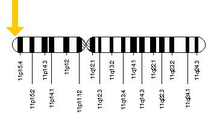
The allele responsible for sickle cell anaemia can be found on the short arm of chromosome 11, more specifically 11p15.5. A person who receives the defective gene from both father and mother develops the disease; a person who receives one defective and one healthy allele remains healthy, but can pass on the disease and is known as a carrier or heterozygote. Heterozygotes are still able to contract malaria, but their symptoms are generally less severe.[41]
Due to the adaptive advantage of the heterozygote, the disease is still prevalent, especially among people with recent ancestry in malaria-stricken areas, such as Africa, the Mediterranean, India, and the Middle East.[42] Malaria was historically endemic to southern Europe, but it was declared eradicated in the mid-20th century, with the exception of rare sporadic cases.[43]
The malaria parasite has a complex lifecycle and spends part of it in red blood cells. In a carrier, the presence of the malaria parasite causes the red blood cells with defective haemoglobin to rupture prematurely, making the Plasmodium parasite unable to reproduce. Further, the polymerization of Hb affects the ability of the parasite to digest Hb in the first place. Therefore, in areas where malaria is a problem, people's chances of survival actually increase if they carry sickle cell trait (selection for the heterozygote).
In the United States, with no endemic malaria, the prevalence of sickle cell anaemia among people of African ancestry is lower (about 0.25%) than among people in West Africa (about 4.0%) and is falling. Without endemic malaria, the sickle cell mutation is purely disadvantageous and tends to decline in the affected population by natural selection, and now artificially through prenatal genetic screening. However, the African American community descends from a significant admixture of several African and non-African ethnic groups and also represents the descendants of survivors of slavery and the slave trade. Thus, a degree of genetic dilution via crossbreeding with non-African people and high health-selective pressure through slavery (especially the slave trade and the frequently deadly Middle Passage) may be the most plausible explanations for the lower prevalence of sickle cell anaemia (and, possibly, other genetic diseases) among African Americans compared to West Africans. Another factor that limits the spread of sickle cell genes in North America is the relative absence of polygamy. In polygamous societies, affected males may father many children with multiple partners.[44]
Pathophysiology

The loss of red blood cell elasticity is central to the pathophysiology of sickle cell disease. Normal red blood cells are quite elastic and have a biconcave disc shape, which allows the cells to deform to pass through capillaries.[45] In sickle cell disease, low oxygen tension promotes red blood cell sickling and repeated episodes of sickling damage the cell membrane and decrease the cell's elasticity. These cells fail to return to normal shape when normal oxygen tension is restored. As a consequence, these rigid blood cells are unable to deform as they pass through narrow capillaries, leading to vessel occlusion and ischaemia.
The actual anaemia of the illness is caused by haemolysis, the destruction of the red cells, because of their shape. Although the bone marrow attempts to compensate by creating new red cells, it does not match the rate of destruction.[46] Healthy red blood cells typically function for 90–120 days, but sickled cells only last 10–20 days.[47]
Diagnosis
In HbS, the complete blood count reveals haemoglobin levels in the range of 6–8 g/dl with a high reticulocyte count (as the bone marrow compensates for the destruction of sickled cells by producing more red blood cells). In other forms of sickle cell disease, Hb levels tend to be higher. A blood film may show features of hyposplenism (target cells and Howell-Jolly bodies).
Sickling of the red blood cells, on a blood film, can be induced by the addition of sodium metabisulfite. The presence of sickle haemoglobin can also be demonstrated with the "sickle solubility test". A mixture of haemoglobin S (HbS) in a reducing solution (such as sodium dithionite) gives a turbid appearance, whereas normal Hb gives a clear solution.
Abnormal haemoglobin forms can be detected on haemoglobin electrophoresis, a form of gel electrophoresis on which the various types of haemoglobin move at varying speeds. Sickle cell haemoglobin (HgbS) and haemoglobin C with sickling (HgbSC)—the two most common forms—can be identified from there. The diagnosis can be confirmed with high-performance liquid chromatography. Genetic testing is rarely performed, as other investigations are highly specific for HbS and HbC.[48]
An acute sickle cell crisis is often precipitated by infection. Therefore, a urinalysis to detect an occult urinary tract infection, and chest X-ray to look for occult pneumonia should be routinely performed.[49]
People who are known carriers of the disease often undergo genetic counseling before they have children. A test to see if an unborn child has the disease takes either a blood sample from the fetus or a sample of amniotic fluid. Since taking a blood sample from a fetus has greater risks, the latter test is usually used. Neonatal screening provides not only a method of early detection for individuals with sickle cell disease, but also allows for identification of the groups of people who carry the sickle cell trait.[50]
Management
Treatment involves a number of measures. While it has been historically recommended that people with sickle cell disease avoid exercise, regular exercise may benefit people.[51] Dehydration should be avoided.[52] A diet high in calcium is recommended[53] but the effectiveness of vitamin D supplementation remains uncertain.[54] L-glutamine use was supported by the FDA starting at the age of five, as it decreases complications.[55]
Folic acid and penicillin
From birth to five years of age, penicillin daily, due to the immature immune system that makes them more prone to early childhood illnesses, is recommended.[56] Dietary supplementation of folic acid had been previously recommended by the WHO.[5] A 2016 Cochrane review of its use found "the effect of supplementation on anaemia and any symptoms of anaemia remains unclear" due to a lack of medical evidence.[57]
Malaria prevention
The protective effect of sickle cell trait does not apply to people with sickle cell disease; in fact, they are more vulnerable to malaria, since the most common cause of painful crises in malarial countries is infection with malaria. People with sickle cell disease living in malarial countries should receive lifelong medication for prevention.[58]
Vaso-occlusive crisis
Most people with sickle cell disease have intensely painful episodes called vaso-occlusive crises. However, the frequency, severity, and duration of these crises vary tremendously. Painful crises are treated symptomatically with pain medications; pain management requires opioid drug administration at regular intervals until the crisis has settled. For milder crises, a subgroup of patients manages on NSAIDs (such as diclofenac or naproxen). For more severe crises, most patients require inpatient management for intravenous opioids; patient-controlled analgesia devices are commonly used in this setting. Diphenhydramine is also an effective agent that doctors frequently prescribe to help control itching associated with the use of opioids.
Extra fluids, administered either orally or intravenously, are a routine part of treatment of vaso-occlusive crises but the evidence about the most effective route, amount and type of fluid replacement remains uncertain.[59]
Crizanlizumab, a monoclonal antibody target towards p-selectin was approved in 2019 in the United States to reduce the frequency of vaso-occlusive crisis in those 16 years and older.[60]
Acute chest syndrome
Management is similar to vaso-occlusive crisis, with the addition of antibiotics (usually a quinolone or macrolide, since cell wall-deficient ["atypical"] bacteria are thought to contribute to the syndrome),[61] oxygen supplementation for hypoxia, and close observation. In the absence of high quality evidence regarding the effectiveness of antibiotics for acute chest syndrome in people with sickle cell disease, there is no standard antibiotic treatment as of 2019.[62] It is recommended that people with suspected acute chest syndrome should be admitted to the hospital with worsening A-a gradient an indication for ICU admission.[25]
Should the pulmonary infiltrate worsen or the oxygen requirements increase, simple blood transfusion or exchange transfusion is indicated. The latter involves the exchange of a significant portion of the person's red cell mass for normal red cells, which decreases the level of haemoglobin S in the patient's blood. However, there is currently uncertain evidence about the possible benefits or harms of blood transfusion for acute chest syndrome in people with sickle cell disease.[63]
Hydroxyurea
Hydroxyurea probably reduces the frequency of painful episodes and the risk of life-threatening illness or death but there is currently insufficient evidence regarding the risk of adverse effects.[64] Hydroxyurea and phlebotomy combined may be more effective than transfusion and chelation combined in terms of pain, life-threatening illness and risk of death.[64]
It was the first approved drug for the treatment of sickle cell anaemia, and was shown to decrease the number and severity of attacks in 1995[65] and shown to possibly increase survival time in a study in 2003.[66] This is achieved, in part, by reactivating fetal haemoglobin production in place of the haemoglobin S that causes sickle cell anaemia. Hydroxyurea had previously been used as a chemotherapy agent, and some concern exists that long-term use may be harmful, but this risk has been shown to be either absent or very small and the benefits likely outweigh the risks.[16][67]
Voxelotor was approved in the United States in 2019 to increase hemoglobin in people with SS disease.[68]
Blood transfusion
Blood transfusions are often used in the management of sickle cell disease in acute cases and to prevent complications by decreasing the number of red blood cells (RBCs) that can sickle by adding normal red blood cells.[69] In children, preventive RBC transfusion therapy has been shown to reduce the risk of first stroke or silent stroke when transcranial Doppler ultrasonography shows abnormal cerebral blood flow.[6] In those who have sustained a prior stroke event, it also reduces the risk of recurrent stroke and additional silent strokes.[70][71]
Bone marrow transplant
Bone marrow transplants have proven effective in children; they are the only known cure for SCD.[72] However, bone marrow transplants are difficult to obtain because of the specific HLA typing necessary. Ideally, a close relative (allogeneic) would donate the bone marrow necessary for transplantation.
Avascular necrosis
When treating avascular necrosis of the bone in people with sickle cell disease, the aim of treatment is to reduce or stop the pain and maintain joint mobility.[73] Current treatment options include rest the joint, physical therapy, pain-relief medicine, joint-replacement surgery, or bone grafting.[73] High quality, randomized, controlled trials are needed to assess the most effective treatment option and determine if a combination of physical therapy and surgery is more effective than physical therapy alone.[73]
Psychological therapies
Psychological therapies such as patient education, cognitive therapy, behavioural therapy, and psychodynamic psychotherapy, that aim to complement current medical treatments, require further research to determine their effectiveness.[21]
Prognosis
About 90% of people survive to age 20, and close to 50% survive beyond age 50.[74] In 2001, according to one study performed in Jamaica, the estimated mean survival for people was 53 years for men and 58 years for women with homozygous SCD.[75] The specific life expectancy in much of the developing world is unknown.[76] In 1975 about 7.3% of people with SCD died before their 23rd birthday; while in 1989 2.6% of people with SCD died by the age of 20.[77]
Complications
Sickle cell anaemia can lead to various complications, including:
- Increased risk of severe bacterial infections is due to loss of functioning spleen tissue (and comparable to the risk of infections after having the spleen removed surgically). These infections are typically caused by encapsulated organisms such as Streptococcus pneumoniae and Haemophilus influenzae. Daily penicillin prophylaxis is the most commonly used treatment during childhood, with some haematologists continuing treatment indefinitely. Patients benefit today from routine vaccination for S. pneumoniae.[78]
- Stroke, which can result from a progressive narrowing of blood vessels, prevents oxygen from reaching the brain. Cerebral infarction occurs in children and cerebral haemorrhage in adults.
- Silent stroke causes no immediate symptoms, but is associated with damage to the brain. Silent stroke is probably five times as common as symptomatic stroke. About 10–15% of children with SCD suffer strokes, with silent strokes predominating in the younger patients.[79][80]
- Cholelithiasis (gallstones) and cholecystitis may result from excessive bilirubin production and precipitation due to prolonged haemolysis.
- Avascular necrosis (aseptic bone necrosis) of the hip and other major joints may occur as a result of ischaemia.[73]
- Decreased immune reactions due to hyposplenism (malfunctioning of the spleen)[81]
- Priapism and infarction of the penis[82]
- Osteomyelitis (bacterial bone infection), the most common cause of osteomyelitis in SCD is Salmonella (especially the atypical serotypes Salmonella typhimurium, Salmonella enteritidis, Salmonella choleraesuis, and Salmonella paratyphi B), followed by Staphylococcus aureus and Gram-negative enteric bacilli perhaps because intravascular sickling of the bowel leads to patchy ischaemic infarction.[83]
- Acute papillary necrosis in the kidneys
- Leg ulcers[84]
- In eyes, background retinopathy, proliferative retinopathy, vitreous haemorrhages, and retinal detachments can result in blindness.[85] Regular annual eye checks are recommended.
- During pregnancy, intrauterine growth retardation, spontaneous abortion, and pre-eclampsia
- Chronic pain: Even in the absence of acute vaso-occlusive pain, many patients have unreported chronic pain.[86]
- Pulmonary hypertension (increased pressure on the pulmonary artery) can lead to strain on the right ventricle and a risk of heart failure; typical symptoms are shortness of breath, decreased exercise tolerance, and episodes of syncope. 21% of children and 30% of adults have evidence of pulmonary hypertension when tested; this is associated with reduced walking distance and increased mortality.[87]
- Chronic kidney failure due to sickle-cell nephropathy manifests itself with hypertension, protein loss in the urine, loss of red blood cells in urine and worsened anaemia. If it progresses to end-stage kidney failure, it carries a poor prognosis.[88]
Epidemiology
The highest frequency of sickle cell disease is found in tropical regions, particularly sub-Saharan Africa, tribal regions of India, and the Middle East.[89] Migration of substantial populations from these high-prevalence areas to low-prevalence countries in Europe has dramatically increased in recent decades and in some European countries, sickle cell disease has now overtaken more familiar genetic conditions such as haemophilia and cystic fibrosis.[90] In 2015, it resulted in about 114,800 deaths.[8]
Sickle cell disease occurs more commonly among people whose ancestors lived in tropical and subtropical sub-Saharan regions where malaria is or was common. Where malaria is common, carrying a single sickle cell allele (trait) confers a heterozygote advantage; humans with one of the two alleles of sickle cell disease show less severe symptoms when infected with malaria.[91]
This condition is inherited in an autosomal recessive pattern, which means both copies of the gene in each cell have mutations. The parents each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition.[92]
Africa
Three-quarters of sickle cell cases occur in Africa. A recent WHO report estimated that around 2% of newborns in Nigeria were affected by sickle cell anaemia, giving a total of 150,000 affected children born every year in Nigeria alone. The carrier frequency ranges between 10 and 40% across equatorial Africa, decreasing to 1–2% on the North African coast and <1% in South Africa.[93] Studies in Africa show a significant decrease in infant mortality rate, ages 2–16 months, because of the sickle cell trait. This happened in areas of predominant malarial cases.[94]
Uganda has the fifth-highest sickle cell disease burden in Africa.[95] One study indicates that 20 000 babies per year are born with sickle cell disease with the sickle cell trait at 13·3% and with disease 0·7%.[96]
United States
The number of people with the disease in the United States is about one in 5,000, mostly affecting Americans of sub-Saharan African descent.[97] In the United States, about one out of 365 African-American children and one in every 16,300 Hispanic-American children have sickle cell anaemia.[98] An estimated 100 thousand Americans have the disease.[98] The life expectancy for men with SCD is approximately 42 years of age while women live approximately six years longer.[99] An additional 2 million are carriers of the sickle cell trait.[100] Most infants with SCD born in the United States are identified by routine neonatal screening. As of 2016 all 50 states include screening for sickle cell disease as part of their newborn screen.[101] The newborn's blood is sampled through a heel-prick and is sent to a lab for testing. The baby must have been eating for a minimum of 24 hours before the heel-prick test can be done. Some states also require a second blood test to be done when the baby is two weeks old to ensure the results.[102] Sickle cell anemia is the most common genetic disorder among African Americans. Approximately 8% are carriers and 1 in 375 are born with the disease.[103] Patient advocates for sickle cell disease have complained that it gets less government and private research funding than similar rare diseases such as cystic fibrosis, with researcher Elliott Vichinsky saying this shows racial discrimination or the role of wealth in health care advocacy.[104]
France
As a result of population growth in African-Caribbean regions of overseas France and immigration from North and sub-Saharan Africa to mainland France, sickle cell disease has become a major health problem in France.[105] SCD has become the most common genetic disease in the country, with an overall birth prevalence of one in 2,415 in metropolitan France, ahead of phenylketonuria (one in 10,862), congenital hypothyroidism (one in 3,132), congenital adrenal hyperplasia (one in 19,008) and cystic fibrosis (one in 5,014) for the same reference period.
Since 2000, neonatal screening of SCD has been performed at national level for all newborns defined as being "at risk" for SCD based on ethnic origin (defined as those born to parents originating from sub-Saharan Africa, North Africa, the Mediterranean area (South Italy, Greece, and Turkey), the Arabic peninsula, the French overseas islands, and the Indian subcontinent).[106]
United Kingdom
In the United Kingdom, between 12,000 and 15,000 people are thought to have sickle cell disease [107] with an estimated 250,000 carriers of the condition in England alone. As the number of carriers is only estimated, all newborn babies in the UK receive a routine blood test to screen for the condition.[108] Due to many adults in high-risk groups not knowing if they are carriers, pregnant women and both partners in a couple are offered screening so they can get counselling if they have the sickle cell trait.[109] In addition, blood donors from those in high-risk groups are also screened to confirm whether they are carriers and whether their blood filters properly.[110] Donors who are found to be carriers are then informed and their blood, while often used for those of the same ethnic group, is not used for those with sickle cell disease who require a blood transfusion.[111]
Middle East
In Saudi Arabia, about 4.2% of the population carry the sickle cell trait and 0.26% have sickle cell disease. The highest prevalence is in the Eastern province, where approximately 17% of the population carry the gene and 1.2% have sickle cell disease.[112] In 2005, Saudi Arabia introduced a mandatory premarital test including HB electrophoresis, which aimed to decrease the incidence of SCD and thalassemia.[113]
In Bahrain, a study published in 1998 that covered about 56,000 people in hospitals in Bahrain found that 2% of newborns have sickle cell disease, 18% of the surveyed people have the sickle cell trait, and 24% were carriers of the gene mutation causing the disease.[114] The country began screening of all pregnant women in 1992 and newborns started being tested if the mother was a carrier. In 2004, a law was passed requiring couples planning to marry to undergo free premarital counseling. These programs were accompanied by public education campaigns.[115]
India and Nepal
Sickle cell disease is common in some ethnic groups of central India,[116] where the prevalence has ranged from 9.4 to 22.2% in endemic areas of Madhya Pradesh, Rajasthan, and Chhattisgarh.[117] It is also endemic among Tharu people of Nepal and India; however, they have a sevenfold lower rate of malaria despite living in a malaria infested zone.[118]
History
The first modern report of sickle cell disease may have been in 1846, where the autopsy of an executed runaway slave was discussed; the key finding was the absence of the spleen.[120][121] Reportedly, African slaves in the United States exhibited resistance to malaria, but were prone to leg ulcers.[121] The abnormal characteristics of the red blood cells, which later lent their name to the condition, was first described by Ernest E. Irons (1877–1959), intern to Chicago cardiologist and professor of medicine James B. Herrick (1861–1954), in 1910. Irons saw "peculiar elongated and sickle-shaped" cells in the blood of a man named Walter Clement Noel, a 20-year-old first-year dental student from Grenada. Noel had been admitted to the Chicago Presbyterian Hospital in December 1904 suffering from anaemia.[13][122] Noel was readmitted several times over the next three years for "muscular rheumatism" and "bilious attacks" but completed his studies and returned to the capital of Grenada (St. George's) to practice dentistry. He died of pneumonia in 1916 and is buried in the Catholic cemetery at Sauteurs in the north of Grenada.[13][14] Shortly after the report by Herrick, another case appeared in the Virginia Medical Semi-Monthly with the same title, "Peculiar Elongated and Sickle-Shaped Red Blood Corpuscles in a Case of Severe Anemia."[123] This article is based on a patient admitted to the University of Virginia Hospital on November 15, 1910.[124] In the later description by Verne Mason in 1922, the name "sickle cell anemia" is first used.[14][125] Childhood problems related to sickle cells disease were not reported until the 1930s, despite the fact that this cannot have been uncommon in African-American populations.[121]
Memphis physician Lemuel Diggs, a prolific researcher into sickle cell disease, first introduced the distinction between sickle cell disease and trait in 1933, although until 1949, the genetic characteristics had not been elucidated by James V. Neel and E.A. Beet.[14] 1949 was the year when Linus Pauling described the unusual chemical behaviour of haemoglobin S, and attributed this to an abnormality in the molecule itself.[14][126] The actual molecular change in HbS was described in the late 1950s by Vernon Ingram.[14] The late 1940s and early 1950s saw further understanding in the link between malaria and sickle cell disease. In 1954, the introduction of haemoglobin electrophoresis allowed the discovery of particular subtypes, such as HbSC disease.[14]
Large-scale natural history studies and further intervention studies were introduced in the 1970s and 1980s, leading to widespread use of prophylaxis against pneumococcal infections amongst other interventions. Bill Cosby's Emmy-winning 1972 TV movie, To All My Friends on Shore, depicted the story of the parents of a child suffering from sickle cell disease.[127] The 1990s had the development of hydroxycarbamide, and reports of cure through bone marrow transplantation appeared in 2007.[14]
Some old texts refer to it as drepanocytosis.[128]
Society and culture
U.S. Social Security
Effective September 15, 2017, the U.S. Social Security Administration issued a Policy Interpretation Ruling providing background information on sickle cell disease and a description of how Social Security evaluates the disease during its adjudication process for disability claims.[129][130]
Stigma in the U.S.
In the U.S., there are stigmas surrounding SCD that discourage people with SCD from receiving necessary care. These stigmas largely affect African Americans, since the majority of cases of SCD in the U.S. are in the African American population.[131] People with SCD experience the impact of stigmas of the disease on multiple aspects of life including social and psychological. Studies have shown that those with SCD frequently feel as though they must keep their diagnosis a secret to avoid discrimination in the workplace and also among peers in relationships.[132] In the 1960s, the US government supported initiatives for workplace screening for genetic diseases in an attempt to be protective towards people with SCD. By having this screening, it was intended that employees would not be placed in environments that could potentially be harmful and trigger SCD.[133] However, in practice, this screening was discriminatory and allowed for employers to place restrictions on employees with SCD and carriers as well.[133]
Furthermore, stigmas also affect access to treatment and medical care. People with SCD require comprehensive treatment to manage the disease and avoid further health complications. The transition between adolescence and adulthood has been shown to be difficult for children with SCD to obtain the necessary treatment for the disease.[134] During this transition, youth are further endangered by multiple factors that can result in more hospitalizations and a growing distrust of the healthcare system. The distrust of the healthcare system is developed by racism in healthcare (See: Racism in Healthcare) towards African-Americans. In the 1970s, many publications by the NIH focused on SCD exclusively in African Americans and encouraged genetic screening for SCD.[133] When Black women presented as being carriers for the disease, they were frequently encouraged by counselors to not have children.[133] The government also did not have regulations on genetic counselors and many at-risk couples were encouraged to have abortions, which was seen as an attempt to limit Black fertility.[133]
Research
Umbilical cord blood transplant
While umbilical cord blood transplant can potentially cure the condition, a suitable donor is available in only 10% of people.[135] About 7% of people also die as a result of the procedure and graft versus host disease may occur.[135]
Gene therapy
In 2001, sickle cell disease reportedly had been successfully treated in mice using gene therapy.[136][137] The researchers used a viral vector to make the mice—which have essentially the same defect that causes human sickle cell disease—express production of fetal haemoglobin (HbF), which an individual normally ceases to produce shortly after birth. In humans, using hydroxyurea to stimulate the production of HbF has been known to temporarily alleviate sickle cell disease symptoms. The researchers demonstrated that this gene therapy method is a more permanent way to increase therapeutic HbF production.[138]
Phase 1 clinical trials of gene therapy for sickle cell disease in humans were started in 2014. The clinical trials will assess the safety of lentiviral vector-modified bone marrow for adults with severe sickle cell disease.[139][140] As of 2018, however, no randomized controlled trials have been reported.[141] A case report for the first person treated was published in March 2017, with a few more people being treated since then.[142][143]
Gene editing platforms like CRISPR/Cas9 have been used to correct the disease-causing mutation in hematopoietic stem cells taken from a person with the condition.[144] In July 2019 the gene-editing tool CRISPR was used to edit bone marrow cells from a person with SCD to "turning on" the gene for fetal haemoglobin.[145]
In 2017 there were twelve clinical trials focusing on gene therapy to treat sickle cell anemia. Of those 12 trials, four of them replaced the mutated HBB gene with a healthy one. Three trials used Mozobil, a medication used to treat types of cancer, to determine whether the increase of stem cells can be used for gene therapy. One trial focused on analyzing bone marrow sample from patients with sickle cell anemia. Another trial experimented with using umbilical cord blood from babies both with and without sickle cell anemia to develop gene therapy.[146]
A Cochrane review designed to examine all the existing randomised clinical trial evidence relating to hematopoietic stem cell transplantation for sickle cell disease found no trials that have been completed and concluded that a well-designed, multicentre randomised controlled trial is needed.[147]
Notes
- Historic numbering put this glutamic acid residue at position 6 due to skipping the methionine (M/Met) start codon in protein amino acid position numbering. Current nomenclature calls for counting the methionine as the first amino acid, resulting in the glutamic acid residue falling at position 7. Many references still refer to position 6 and both should likely be referenced for clarity.
References
- "What Are the Signs and Symptoms of Sickle Cell Disease?". National Heart, Lung, and Blood Institute. June 12, 2015. Archived from the original on 9 March 2016. Retrieved 8 March 2016.
- "What Is Sickle Cell Disease?". National Heart, Lung, and Blood Institute. June 12, 2015. Archived from the original on 6 March 2016. Retrieved 8 March 2016.
- "What Causes Sickle Cell Disease?". National Heart, Lung, and Blood Institute. June 12, 2015. Archived from the original on 24 March 2016. Retrieved 8 March 2016.
- "How Is Sickle Cell Disease Diagnosed?". National Heart, Lung, and Blood Institute. June 12, 2015. Archived from the original on 9 March 2016. Retrieved 8 March 2016.
- "Sickle-cell disease and other haemoglobin disorders Fact sheet N°308". January 2011. Archived from the original on 9 March 2016. Retrieved 8 March 2016.
- "How Is Sickle Cell Disease Treated?". National Heart, Lung, and Blood Institute. June 12, 2015. Archived from the original on 9 March 2016. Retrieved 8 March 2016.
- GBD 2015 Disease Injury Incidence Prevalence Collaborators (October 2016). "Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015". Lancet. 388 (10053): 1545–1602. doi:10.1016/S0140-6736(16)31678-6. PMC 5055577. PMID 27733282.
- GBD 2015 Mortality Causes of Death Collaborators (October 2016). "Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015". Lancet. 388 (10053): 1459–1544. doi:10.1016/S0140-6736(16)31012-1. PMC 5388903. PMID 27733281.
- "Learning About Sickle Cell Disease". National Human Genome Research Institute. May 9, 2016. Archived from the original on January 4, 2017. Retrieved January 23, 2017.
- Global Burden of Disease Study 2013 Collaborators (August 2015). "Global, regional, and national incidence, prevalence, and years lived with disability for 301 acute and chronic diseases and injuries in 188 countries, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013". Lancet. 386 (9995): 743–800. doi:10.1016/s0140-6736(15)60692-4. PMC 4561509. PMID 26063472.
- Rees DC, Williams TN, Gladwin MT (December 2010). "Sickle-cell disease". Lancet. 376 (9757): 2018–31. doi:10.1016/s0140-6736(10)61029-x. PMID 21131035.
- Elzouki, Abdelaziz Y. (2012). Textbook of clinical pediatrics (2 ed.). Berlin: Springer. p. 2950. ISBN 9783642022012.
- Savitt TL, Goldberg MF (January 1989). "Herrick's 1910 case report of sickle cell anemia. The rest of the story". JAMA. 261 (2): 266–71. doi:10.1001/jama.261.2.266. PMID 2642320.
- Serjeant GR (December 2010). "One hundred years of sickle cell disease". British Journal of Haematology. 151 (5): 425–9. doi:10.1111/j.1365-2141.2010.08419.x. PMID 20955412.
- National Library of Medicine. URL = ghr.nlm.nih.gov/condition/sickle-cell-disease
- Yawn BP, Buchanan GR, Afenyi-Annan AN, Ballas SK, Hassell KL, James AH, Jordan L, Lanzkron SM, Lottenberg R, Savage WJ, Tanabe PJ, Ware RE, Murad MH, Goldsmith JC, Ortiz E, Fulwood R, Horton A, John-Sowah J (September 2014). "Management of sickle cell disease: summary of the 2014 evidence-based report by expert panel members". JAMA. 312 (10): 1033–48. doi:10.1001/jama.2014.10517. PMID 25203083. S2CID 37681044.
- "BestBets: How long should an average sickle cell crisis last?". Archived from the original on 2010-06-17. Retrieved 2010-11-27.
- Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; Aster, Jon (2009-05-28). Robbins and Cotran Pathologic Basis of Disease, Professional Edition: Expert Consult – Online (Robbins Pathology) (Kindle Locations 33498-33499). Elsevier Health. Kindle Edition.
- Olujohungbe A, Burnett AL (March 2013). "How I manage priapism due to sickle cell disease". British Journal of Haematology. 160 (6): 754–65. doi:10.1111/bjh.12199. PMID 23293942.
- Glassberg J (August 2011). "Evidence-based management of sickle cell disease in the emergency department". Emergency Medicine Practice. 13 (8): 1–20, quiz 20. PMID 22164362.
- Anie KA, Green J (May 2015). "Psychological therapies for sickle cell disease and pain". The Cochrane Database of Systematic Reviews (5): CD001916. doi:10.1002/14651858.CD001916.pub3. PMC 7063720. PMID 25966336.
- Pearson HA (August 1977). "Sickle cell anemia and severe infections due to encapsulated bacteria" (Free full text). The Journal of Infectious Diseases. 136 Suppl: S25–30. doi:10.1093/infdis/136.Supplement.S25. PMID 330779. Archived from the original on 2016-05-27.
- Wong WY, Powars DR, Chan L, Hiti A, Johnson C, Overturf G (March 1992). "Polysaccharide encapsulated bacterial infection in sickle cell anemia: a thirty year epidemiologic experience". American Journal of Hematology. 39 (3): 176–82. doi:10.1002/ajh.2830390305. PMID 1546714.
- Khatib R, Rabah R, Sarnaik SA (January 2009). "The spleen in the sickling disorders: an update". Pediatric Radiology. 39 (1): 17–22. doi:10.1007/s00247-008-1049-9. PMID 19002450. S2CID 2547649.
- Glassberg J (August 2011). "Evidence-based management of sickle cell disease in the emergency department". Emergency Medicine Practice. 13 (8): 1–20, quiz 20. PMID 22164362.
- Mekontso Dessap A, Leon R, Habibi A, Nzouakou R, Roudot-Thoraval F, Adnot S, Godeau B, Galacteros F, Brun-Buisson C, Brochard L, Maitre B (March 2008). "Pulmonary hypertension and cor pulmonale during severe acute chest syndrome in sickle cell disease". American Journal of Respiratory and Critical Care Medicine. 177 (6): 646–53. CiteSeerX 10.1.1.504.790. doi:10.1164/rccm.200710-1606OC. PMID 18174543.
- Paul RN, Castro OL, Aggarwal A, Oneal PA (September 2011). "Acute chest syndrome: sickle cell disease". European Journal of Haematology. 87 (3): 191–207. doi:10.1111/j.1600-0609.2011.01647.x. PMID 21615795.
- Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; Aster, Jon (2009-05-28). Robbins and Cotran Pathologic Basis of Disease, Professional Edition: Expert Consult – Online (Robbins Pathology) (Kindle Location 33329). Elsevier Health. Kindle Edition.
- Slavov SN, Kashima S, Pinto AC, Covas DT (August 2011). "Human parvovirus B19: general considerations and impact on patients with sickle-cell disease and thalassemia and on blood transfusions". FEMS Immunology and Medical Microbiology. 62 (3): 247–62. doi:10.1111/j.1574-695X.2011.00819.x. PMID 21585562.
- Balgir RS (March 2012). "Community expansion and gene geography of sickle cell trait and G6PD deficiency, and natural selection against malaria: experience from tribal land of India". Cardiovascular & Hematological Agents in Medicinal Chemistry. 10 (1): 3–13. doi:10.2174/187152512799201190. PMID 22264009.
- Jadavji T, Prober CG (April 1985). "Dactylitis in a child with sickle cell trait". Canadian Medical Association Journal. 132 (7): 814–5. PMC 1345873. PMID 3978504.
- Worrall VT, Butera V (December 1976). "Sickle-cell dactylitis". The Journal of Bone and Joint Surgery. American Volume. 58 (8): 1161–3. doi:10.2106/00004623-197658080-00024. PMID 1002763. Archived from the original on 2016-09-23.
- Miller ST (May 2011). "How I treat acute chest syndrome in children with sickle cell disease". Blood. 117 (20): 5297–305. doi:10.1182/blood-2010-11-261834. PMID 21406723. S2CID 206896811.
- James, William D.; Berger, Timothy G.; et al. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. p. 847. ISBN 978-0-7216-2921-6.
- Sankaran VG, Orkin SH (January 2013). "The switch from fetal to adult hemoglobin". Cold Spring Harbor Perspectives in Medicine. 3 (1): a011643. doi:10.1101/cshperspect.a011643. PMC 3530042. PMID 23209159.
- "Sickle Cell Disease". NORD (National Organization for Rare Disorders). Retrieved 10 June 2019.
- "sickle cell disease". Genetics Home Reference. Archived from the original on 2016-05-15. Retrieved 2016-05-07.
- Green NS, Fabry ME, Kaptue-Noche L, Nagel RL (October 1993). "Senegal haplotype is associated with higher HbF than Benin and Cameroon haplotypes in African children with sickle cell anemia". American Journal of Hematology. 44 (2): 145–6. doi:10.1002/ajh.2830440214. PMID 7505527.
- Suzanne Clancy (2008). "Genetic mutation". Nature Education. 1 (1): 187.
- Wellstein A, Pitschner HF (July 1988). "Complex dose-response curves of atropine in man explained by different functions of M1- and M2-cholinoceptors". Naunyn-Schmiedeberg's Archives of Pharmacology. 338 (1): 19–27. doi:10.1007/bf00168807. PMC 3237253. PMID 22089617.
- Allison AC (October 2009). "Genetic control of resistance to human malaria". Current Opinion in Immunology. 21 (5): 499–505. doi:10.1016/j.coi.2009.04.001. PMID 19442502.
- Kwiatkowski DP (August 2005). "How malaria has affected the human genome and what human genetics can teach us about malaria". American Journal of Human Genetics. 77 (2): 171–92. doi:10.1086/432519. PMC 1224522. PMID 16001361.
- Ponçon N, Toty C, L'Ambert G, Le Goff G, Brengues C, Schaffner F, Fontenille D (February 2007). "Biology and dynamics of potential malaria vectors in Southern France". Malaria Journal. 6 (1): 18. doi:10.1186/1475-2875-6-18. PMC 1808464. PMID 17313664.
- Lesi FE, Bassey EE (July 1972). "Family study in sickle cell disease in Nigeria". Journal of Biosocial Science. 4 (3): 307–13. doi:10.1017/S0021932000008622. PMID 5041262.
- Capriotti, Theresa (2016). Pathophysiology : introductory concepts and clinical perspectives. Frizzell, Joan Parker. Philadelphia. ISBN 9780803615717. OCLC 900626405.
- "How Does Sickle Cell Cause Disease?". Archived from the original on 2010-09-23. Retrieved 2010-11-27.
- "Sickle Cell Anemia: eMedicine Emergency Medicine". Archived from the original on 2010-12-04. Retrieved 2010-11-27.
- Clarke GM, Higgins TN (August 2000). "Laboratory investigation of hemoglobinopathies and thalassemias: review and update". Clinical Chemistry. 46 (8 Pt 2): 1284–90. doi:10.1093/clinchem/46.8.1284. PMID 10926923. Archived from the original on 2008-03-20.
- "BestBets: Does routine urinalysis and chest radiography detect occult bacterial infection in sickle cell patients presenting to the accident and emergency department with painful crisis?". Archived from the original on 2010-06-17. Retrieved 2010-11-27.
- Lee, C., Davies, S.,& Dezatoux, C. (2000). Neonatal Screening for sickle cell disease. The Cochrane Collaboration. John Wiley & Sons, Ltd.
- Martin, Cyril; Pialoux, Vincent; Faes, Camille; Charrin, Emmanuelle; Skinner, Sarah; Connes, Philippe (February 2018). "Does physical activity increase or decrease the risk of sickle cell disease complications?". British Journal of Sports Medicine. 52 (4): 214–218. doi:10.1136/bjsports-2015-095317. PMID 26701924. S2CID 24464344.
- "Keeping Well with Sickle Cell Disease - Brent Sickle Cell & Thalassaemia Centre". www.sickle-thal.nwlh.nhs.uk. Retrieved 4 October 2019.
- "Nutrition for the Child with Sickle Cell Anemia". www.eatright.org. Retrieved 5 October 2019.
- Soe, Htoo Htoo Kyaw; Abas, Adinegara Bl; Than, Nan Nitra; Ni, Han; Singh, Jaspal; Said, Abdul Razzak Bin Mohd; Osunkwo, Ifeyinwa (28 May 2020). "Vitamin D supplementation for sickle cell disease". The Cochrane Database of Systematic Reviews. 5: CD010858. doi:10.1002/14651858.CD010858.pub3. ISSN 1469-493X. PMID 32462740.
- Commissioner, Office of the (7 July 2017). "Press Announcements – FDA approves new treatment for sickle cell disease". www.fda.gov. Archived from the original on 10 July 2017. Retrieved 10 July 2017.
- "Evidence-Based Management of Sickle Cell Disease" (PDF). 2014. Retrieved November 16, 2017.
twice-daily prophylactic penicillin beginning in early infancy and continuing through at least age 5
- Dixit R, Nettem S, Madan SS, Soe HH, Abas AB, Vance LD, Stover PJ (March 2018). "Folate supplementation in people with sickle cell disease". The Cochrane Database of Systematic Reviews. 3: CD011130. doi:10.1002/14651858.CD011130.pub3. PMC 5440187. PMID 29546732.
- Oniyangi, Oluseyi; Omari, Aika Aa (2019). "Malaria chemoprophylaxis in sickle cell disease". The Cochrane Database of Systematic Reviews. 2019 (11). doi:10.1002/14651858.CD003489.pub2. ISSN 1469-493X. PMC 6532723. PMID 31681984.
- Okomo, U; Meremikwu, MM (31 July 2017). "Fluid replacement therapy for acute episodes of pain in people with sickle cell disease". The Cochrane Database of Systematic Reviews. 7: CD005406. doi:10.1002/14651858.CD005406.pub5. PMC 6483538. PMID 28759112.
- Research, Center for Drug Evaluation and (2019-12-20). "FDA approves crizanlizumab-tmca for sickle cell disease". FDA.
- Aldrich TK, Nagel RL (1998). "Pulmonary Complications of Sickle Cell Disease.". In Reynolds HY, Bone RC, Dantzker DR, George RB, Matthay RA (eds.). Pulmonary and Critical Care Medicine (6th ed.). St. Louis: Mosby. pp. 1–10. ISBN 978-0-8151-1371-3.
- Martí-Carvajal, AJ; Conterno, LO; Knight-Madden, JM (18 September 2019). "Antibiotics for treating acute chest syndrome in people with sickle cell disease". The Cochrane Database of Systematic Reviews. 9: CD006110. doi:10.1002/14651858.CD006110.pub5. PMC 6749554. PMID 31531967.
- Dolatkhah, R; Dastgiri, S (16 January 2020). "Blood transfusions for treating acute chest syndrome in people with sickle cell disease". The Cochrane Database of Systematic Reviews. 1: CD007843. doi:10.1002/14651858.CD007843.pub4. PMC 6984655. PMID 31942751.
- Nevitt, SJ; Jones, AP; Howard, J (20 April 2017). "Hydroxyurea (hydroxycarbamide) for sickle cell disease". The Cochrane Database of Systematic Reviews. 4: CD002202. doi:10.1002/14651858.CD002202.pub2. PMC 6478259. PMID 28426137.
- Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP, Bonds DR (May 1995). "Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia". The New England Journal of Medicine. 332 (20): 1317–22. doi:10.1056/NEJM199505183322001. PMID 7715639.
- Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, Orringer E, Bellevue R, Olivieri N, Eckman J, Varma M, Ramirez G, Adler B, Smith W, Carlos T, Ataga K, DeCastro L, Bigelow C, Saunthararajah Y, Telfer M, Vichinsky E, Claster S, Shurin S, Bridges K, Waclawiw M, Bonds D, Terrin M (April 2003). "Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment". JAMA. 289 (13): 1645–51. doi:10.1001/jama.289.13.1645. PMID 12672732.
- Platt OS (March 2008). "Hydroxyurea for the treatment of sickle cell anemia". The New England Journal of Medicine. 358 (13): 1362–9. doi:10.1056/NEJMct0708272. PMID 18367739. S2CID 351061.
- Research, Center for Drug Evaluation and (25 November 2019). "FDA approves voxelotor for sickle cell disease". FDA. Retrieved 9 December 2019.
- Drasar E, Igbineweka N, Vasavda N, Free M, Awogbade M, Allman M, Mijovic A, Thein SL (March 2011). "Blood transfusion usage among adults with sickle cell disease - a single institution experience over ten years". British Journal of Haematology. 152 (6): 766–70. doi:10.1111/j.1365-2141.2010.08451.x. PMID 21275951.
- Gyang E, Yeom K, Hoppe C, Partap S, Jeng M (January 2011). "Effect of chronic red cell transfusion therapy on vasculopathies and silent infarcts in patients with sickle cell disease". American Journal of Hematology. 86 (1): 104–6. doi:10.1002/ajh.21901. PMID 21117059.
- Mirre E, Brousse V, Berteloot L, Lambot-Juhan K, Verlhac S, Boulat C, Dumont MD, Lenoir G, de Montalembert M (March 2010). "Feasibility and efficacy of chronic transfusion for stroke prevention in children with sickle cell disease". European Journal of Haematology. 84 (3): 259–65. doi:10.1111/j.1600-0609.2009.01379.x. PMID 19912310.
- Walters MC, Patience M, Leisenring W, Eckman JR, Scott JP, Mentzer WC, Davies SC, Ohene-Frempong K, Bernaudin F, Matthews DC, Storb R, Sullivan KM (August 1996). "Bone marrow transplantation for sickle cell disease". The New England Journal of Medicine. 335 (6): 369–76. doi:10.1056/NEJM199608083350601. PMID 8663884. S2CID 25256772.
- Martí-Carvajal, Arturo J.; Solà, Ivan; Agreda-Pérez, Luis H. (2019). "Treatment for avascular necrosis of bone in people with sickle cell disease". The Cochrane Database of Systematic Reviews. 12: CD004344. doi:10.1002/14651858.CD004344.pub7. ISSN 1469-493X. PMC 6894369. PMID 31803937.
- Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; Aster, Jon (2009-05-28). Robbins and Cotran Pathologic Basis of Disease, Professional Edition: Expert Consult – Online (Robbins Pathology) (Kindle Locations 33530-33531). Elsevier Health. Kindle Edition.
- Wierenga KJ, Hambleton IR, Lewis NA (March 2001). "Survival estimates for patients with homozygous sickle-cell disease in Jamaica: a clinic-based population study". Lancet. 357 (9257): 680–3. doi:10.1016/s0140-6736(00)04132-5. PMID 11247552.
- Costa FF, Conran N (2016). Sickle Cell Anemia: From Basic Science to Clinical Practice. Springer. p. 35. ISBN 9783319067131. Retrieved 8 May 2016.
- Prabhakar, H; Haywood C, Jr; Molokie, R (May 2010). "Sickle cell disease in the United States: looking back and forward at 100 years of progress in management and survival". American Journal of Hematology. 85 (5): 346–53. doi:10.1002/ajh.21676. PMID 20425797.
- Kavanagh PL, Sprinz PG, Vinci SR, Bauchner H, Wang CJ (December 2011). "Management of children with sickle cell disease: a comprehensive review of the literature". Pediatrics. 128 (6): e1552–74. doi:10.1542/peds.2010-3686. PMID 22123880. S2CID 14524078. Archived from the original on 2016-03-04.
- Adams RJ, Ohene-Frempong K, Wang W (2001). "Sickle cell and the brain". Hematology. American Society of Hematology. Education Program. 2001 (1): 31–46. doi:10.1182/asheducation-2001.1.31. PMID 11722977.
- Adams RJ (November 2007). "Big strokes in small persons". Archives of Neurology. 64 (11): 1567–74. doi:10.1001/archneur.64.11.1567. PMID 17998439.
- Kenny MW, George AJ, Stuart J (July 1980). "Platelet hyperactivity in sickle-cell disease: a consequence of hyposplenism". Journal of Clinical Pathology. 33 (7): 622–5. doi:10.1136/jcp.33.7.622. PMC 1146172. PMID 7430367.
- Chrouser KL, Ajiboye OB, Oyetunji TA, Chang DC (April 2011). "Priapism in the United States: the changing role of sickle cell disease". American Journal of Surgery. 201 (4): 468–74. doi:10.1016/j.amjsurg.2010.03.017. PMID 21421100.
- Almeida A, Roberts I (May 2005). "Bone involvement in sickle cell disease". British Journal of Haematology. 129 (4): 482–90. doi:10.1111/j.1365-2141.2005.05476.x. PMID 15877730. Archived from the original on 2012-12-16.
- Rudge FW (1991). "Hyperbaric oxygen therapy in the treatment of sickle cell leg ulcers". J. Hyperbaric Med. 6 (1): 1–4. Retrieved 2011-03-23.
- Elagouz M, Jyothi S, Gupta B, Sivaprasad S (July 2010). "Sickle cell disease and the eye: old and new concepts". Survey of Ophthalmology. 55 (4): 359–77. doi:10.1016/j.survophthal.2009.11.004. PMID 20452638.
- Smith WR, Penberthy LT, Bovbjerg VE, McClish DK, Roberts JD, Dahman B, Aisiku IP, Levenson JL, Roseff SD (January 2008). "Daily assessment of pain in adults with sickle cell disease". Annals of Internal Medicine. 148 (2): 94–101. CiteSeerX 10.1.1.690.5870. doi:10.7326/0003-4819-148-2-200801150-00004. PMID 18195334. S2CID 34924760.
- Caughey MC, Poole C, Ataga KI, Hinderliter AL (August 2015). "Estimated pulmonary artery systolic pressure and sickle cell disease: a meta-analysis and systematic review". British Journal of Haematology. 170 (3): 416–24. doi:10.1111/bjh.13447. PMID 25854714.
- Powars DR, Elliott-Mills DD, Chan L, Niland J, Hiti AL, Opas LM, Johnson C (October 1991). "Chronic renal failure in sickle cell disease: risk factors, clinical course, and mortality". Annals of Internal Medicine. 115 (8): 614–20. doi:10.7326/0003-4819-115-8-614. PMID 1892333.
- Weatherall DJ, Clegg JB (2001). "Inherited haemoglobin disorders: an increasing global health problem". Bulletin of the World Health Organization. 79 (8): 704–12. PMC 2566499. PMID 11545326.
- Roberts I, de Montalembert M (July 2007). "Sickle cell disease as a paradigm of immigration hematology: new challenges for hematologists in Europe". Haematologica. 92 (7): 865–71. doi:10.3324/haematol.11474. PMID 17606434.
- Wellems TE, Hayton K, Fairhurst RM (September 2009). "The impact of malaria parasitism: from corpuscles to communities". The Journal of Clinical Investigation. 119 (9): 2496–505. doi:10.1172/JCI38307. PMC 2735907. PMID 19729847.
- National Library of Medicine. URL = https://ghr.nlm.nih.gov/condition/sickle-cell-disease#statistics Archived 2016-05-15 at the Wayback Machine
- WHO. "Sickle-cell anaemia – Report by the Secretariat" (PDF). Archived from the original (PDF) on 2011-01-04. Retrieved 2010-11-27.
- Aidoo M, Terlouw DJ, Kolczak MS, McElroy PD, ter Kuile FO, Kariuki S, Nahlen BL, Lal AA, Udhayakumar V (April 2002). "Protective effects of the sickle cell gene against malaria morbidity and mortality". Lancet. 359 (9314): 1311–2. doi:10.1016/S0140-6736(02)08273-9. PMID 11965279. S2CID 37952036.
- Tusuubira, Sharifu K.; Nakayinga, Ritah; Mwambi, Bashir; Odda, John; Kiconco, Sylvia; Komuhangi, Alimah (27 April 2018). "Knowledge, perception and practices towards sickle cell disease: a community survey among adults in Lubaga division, Kampala Uganda". BMC Public Health. 18 (1): 561. doi:10.1186/s12889-018-5496-4. PMC 5924488. PMID 29703184.
- Ndeezi, Grace; Kiyaga, Charles; Hernandez, Arielle G; Munube, Deogratias; Howard, Thad A; Ssewanyana, Isaac; Nsungwa, Jesca; Kiguli, Sarah; Ndugwa, Christopher M; Ware, Russell E; Aceng, Jane R (March 2016). "Burden of sickle cell trait and disease in the Uganda Sickle Surveillance Study (US3): a cross-sectional study". The Lancet Global Health. 4 (3): e195–e200. doi:10.1016/S2214-109X(15)00288-0. PMID 26833239.
- National Heart, Lung and Blood Institute. "Sickle cell anemia, key points". Archived from the original on 2010-12-02. Retrieved 2010-11-27.
- "Data & Statistics on Sickle Cell Disease | CDC". Centers for Disease Control and Prevention. 31 August 2016. Retrieved 13 December 2019.
- "September is Sickle Cell Awareness Month". CDC. Archived from the original on 27 September 2010. Retrieved 6 February 2011.
- "Sickle Cell Trait". www.hematology.org. 8 September 2017. Retrieved 13 December 2019.
- "Disorder Name: Sickle Cell Disease". New Born Screening. Archived from the original on 28 September 2016. Retrieved 11 October 2016.
- "default - Stanford Children's Health". www.stanfordchildrens.org. Retrieved 2020-03-14.
- Edwards, Q. T.; Seibert, D.; Macri, C.; Carolyn, C.; Tilghman, J. (November 2004). "Assessing ethnicity in preconception counseling: Genetics--what nurse practitioners need to know". Clinical Practice. 16 (11): 472–480. doi:10.1111/j.1745-7599.2004.tb00426.x. PMID 15617360. S2CID 7644129.
- "Sickle Cell Patients Endure Discrimination, Poor Care And Shortened Lives". NPR.org. November 4, 2017. Retrieved 12 November 2017.
- Bardakdjian J, Wajcman H (September 2004). "[Epidemiology of sickle cell anemia]". La Revue du Praticien (in French). 54 (14): 1531–3. PMID 15558961.
- Thuret I, Sarles J, Merono F, Suzineau E, Collomb J, Lena-Russo D, Levy N, Bardakdjian J, Badens C (June 2010). "Neonatal screening for sickle cell disease in France: evaluation of the selective process". Journal of Clinical Pathology. 63 (6): 548–51. doi:10.1136/jcp.2009.068874. PMID 20498028. S2CID 22391674.
- "Inheriting sickle cell anaemia – Live Well – NHS Choices". www.nhs.uk. 2017-10-23. Archived from the original on 2014-12-02.
- "Sickle cell anaemia – NHS Choices". www.nhs.uk. 2017-10-23. Archived from the original on 2011-12-13.
- "Who is offered screening and when?". screening.nhs.uk. Archived from the original on 2014-12-31.
- "Give Blood – Resources – Sickle Cell and Blood Donation". Give Blood. Archived from the original on 2014-12-31.
- "Why is Blood from Afro-Caribbean Donors Special?". sicklecellsociety.org. Archived from the original on 2014-12-30.
- Jastaniah W (2011). "Epidemiology of sickle cell disease in Saudi Arabia". Annals of Saudi Medicine. 31 (3): 289–93. doi:10.4103/0256-4947.81540. PMC 3119971. PMID 21623060.
- Memish ZA, Saeedi MY (2011). "Six-year outcome of the national premarital screening and genetic counseling program for sickle cell disease and β-thalassemia in Saudi Arabia". Annals of Saudi Medicine. 31 (3): 229–35. doi:10.4103/0256-4947.81527. PMC 3119961. PMID 21623050.
- Al Arrayed, Sheikha (1995). "Features of sickle-cell disease in Bahrain". Eastern Mediterranean Health Journal. 1 (1). Archived from the original on 2016-10-08.
- Al Arrayed S, Al Hajeri A (2010). "Public awareness of sickle cell disease in Bahrain". Annals of Saudi Medicine. 30 (4): 284–8. doi:10.4103/0256-4947.65256. PMC 2931779. PMID 20622345.
- "Sickle Cell Anemia". www.hematology.org. 2014-12-16. Archived from the original on 2017-06-25. Retrieved 2017-05-01.
- Awasthy N, Aggarwal KC, Goyal PC, Prasad MS, Saluja S, Sharma M (2008). "Sickle cell disease: Experience of a tertiary care center in a nonendemic area". Annals of Tropical Medicine and Public Health. 1 (1): 1–4. doi:10.4103/1755-6783.43069.
- "Life with sickle cell – Nation – Nepali Times". Archived from the original on 2015-06-24.
- Asnani MR, McCaw-Binns AM, Reid ME (2011). "Excess risk of maternal death from sickle cell disease in Jamaica: 1998-2007". PLOS ONE. 6 (10): e26281. Bibcode:2011PLoSO...626281A. doi:10.1371/journal.pone.0026281. PMC 3200316. PMID 22039456.
- Lebby R (1846). "Case of absence of the spleen". Southern J of Med Pharmacol. 1: 481–3.
- Ballas SK, Gupta K, Adams-Graves P (November 2012). "Sickle cell pain: a critical reappraisal". Blood. 120 (18): 3647–56. doi:10.1182/blood-2012-04-383430. PMID 22923496.
- Herrick JB (1 November 1910). "Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia". Archives of Internal Medicine. 6 (5): 179–184. doi:10.1001/archinte.1910.00050330050003.; reprinted as Herrick JB (2001). "Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. 1910". The Yale Journal of Biology and Medicine. 74 (3): 179–84. PMC 2588723. PMID 11501714.
- Washburn RE (1911). "Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia". The Virginia Medical Semi-Monthly. 15 (21): 490–493.
- "UVa Hospital Celebrating 100 Years". University of Virginia. Archived from the original on 31 January 2015. Retrieved 28 January 2015.
- Mason VR (1922). "Sickle cell anemia". JAMA. 79 (16): 1318–1320. doi:10.1001/jama.1922.02640160038012. Reprinted in PMID 3900438
- Pauling L, Itano HA (November 1949). "Sickle cell anemia a molecular disease". Science. 110 (2865): 543–8. Bibcode:1949Sci...110..543P. doi:10.1126/science.110.2865.543. PMID 15395398.
- "Foster, Gloria". Facts On File History Database. Archived from the original on 2016-03-05. Retrieved 2015-02-25.
- Richard-Lenoble D, Toublanc JE, Zinsou RD, Kombila M, Carme B (1980). "Résultats de l'étude systématique de la drépanocytose par électrophorèse de l'hémoglobine chez 1500 gabonais" [Results of a systematic study of drepanocytosis in 1,500 Gabonese using hemoglobin electrophoresis]. Bulletin de la Societe de Pathologie Exotique et de Ses Filiales (in French). 73 (2): 200–6. PMID 7460122.
- SSA, Office of Disability Policy. "Social Security Ruling: SSR 2017-3p". www.ssa.gov. Retrieved 2018-01-15.
- "Federal Register, Volume 82 Issue 178 (Friday, September 15, 2017)". www.gpo.gov. Retrieved 2018-01-15.
- "Sickle Cell Disease | National Heart, Lung, and Blood Institute (NHLBI)". www.nhlbi.nih.gov. Retrieved 2020-07-04.
- Bulgin, Dominique; Tanabe, Paula; Jenerette, Coretta (2018). "Stigma of Sickle Cell Disease: A Systematic Review". Issues in Mental Health Nursing. 39 (8): 675–686. doi:10.1080/01612840.2018.1443530. ISSN 0161-2840. PMC 6186193. PMID 29652215.
- Washington, Harriet A. (2006). Medical apartheid : the dark history of medical experimentation on Black Americans from colonial times to the present (1st paperback ed.). New York: Harlem Moon. ISBN 978-0-7679-1547-2. OCLC 192050177.
- Jenerette, Coretta M.; Brewer, Cheryl (2010). "Health-Related Stigma in Young Adults With Sickle Cell Disease". Journal of the National Medical Association. 102 (11): 1050–1055. doi:10.1016/s0027-9684(15)30732-x. ISSN 0027-9684. PMC 3119340. PMID 21141294.
- Kassim AA, Sharma D (December 2017). "Hematopoietic stem cell transplantation for sickle cell disease: The changing landscape". Hematology/Oncology and Stem Cell Therapy. 10 (4): 259–266. doi:10.1016/j.hemonc.2017.05.008. PMID 28641096.
- Pawliuk R, Westerman KA, Fabry ME, Payen E, Tighe R, Bouhassira EE, Acharya SA, Ellis J, London IM, Eaves CJ, Humphries RK, Beuzard Y, Nagel RL, Leboulch P (December 2001). "Correction of sickle cell disease in transgenic mouse models by gene therapy". Science. 294 (5550): 2368–71. Bibcode:2001Sci...294.2368P. doi:10.1126/science.1065806. PMID 11743206. S2CID 25607771.
- Wilson JF (18 March 2002). "Murine Gene Therapy Corrects Symptoms of Sickle Cell Disease". The Scientist – Magazine of the Life Sciences. Retrieved 17 December 2014.
- St. Jude Children's Research Hospital (4 December 2008). "Gene Therapy Corrects Sickle Cell Disease In Laboratory Study". ScienceDaily. Archived from the original on 13 December 2014. Retrieved 17 December 2014.
- Clinical trial number NCT02247843 for "Stem Cell Gene Therapy for Sickle Cell Disease" at ClinicalTrials.gov
- Clinical trial number NCT00012545 for "Collection and Storage of Umbilical Cord Stem Cells for Treatment of Sickle Cell Disease" at ClinicalTrials.gov
- Olowoyeye A, Okwundu CI (November 2018). "Gene therapy for sickle cell disease". The Cochrane Database of Systematic Reviews. 11: CD007652. doi:10.1002/14651858.CD007652.pub6. PMC 6517046. PMID 30480767.
- Ribeil JA, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, Caccavelli L, Neven B, Bourget P, El Nemer W, Bartolucci P, Weber L, Puy H, Meritet JF, Grevent D, Beuzard Y, Chrétien S, Lefebvre T, Ross RW, Negre O, Veres G, Sandler L, Soni S, de Montalembert M, Blanche S, Leboulch P, Cavazzana M (March 2017). "Gene Therapy in a Patient with Sickle Cell Disease". The New England Journal of Medicine. 376 (9): 848–855. doi:10.1056/NEJMoa1609677. PMID 28249145. S2CID 5128871.
- Kolata G (27 January 2019). "These Patients Had Sickle-Cell Disease. Experimental Therapies Might Have Cured Them". The New York Times. Retrieved 28 January 2019.
- Dever, Daniel P.; Bak, Rasmus O.; Reinisch, Andreas; Camarena, Joab; Washington, Gabriel; Nicolas, Carmencita E.; Pavel-Dinu, Mara; Saxena, Nivi; Wilkens, Alec B. (November 17, 2016). "CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells". Nature. 539 (7629): 384–389. Bibcode:2016Natur.539..384D. doi:10.1038/nature20134. ISSN 1476-4687. PMC 5898607. PMID 27820943.
- "In A 1st, Doctors In U.S. Use CRISPR Tool To Treat Patient With Genetic Disorder". NPR.org. Retrieved 2019-07-31.
- Walker, Meredith (2018-01-15). "Gene Therapy". Sickle Cell Disease News. Retrieved 2020-03-14.
- Oringanje, C; Nemecek, E; Oniyangi, O (3 July 2020). "Hematopoietic stem cell transplantation for people with sickle cell disease". The Cochrane database of systematic reviews. 7: CD007001. doi:10.1002/14651858.CD007001.pub5. PMID 32617981.
Further reading
| Library resources about Sickle cell disease |
- Brown, Robert T., ed. (2006). Comprehensive handbook of childhood cancer and sickle cell disease: a biopsychosocial approach. Oxford University Press. ISBN 978-0-19-516985-0.
- Hill, Shirley A. (2003). Managing Sickle Cell Disease in Low-Income Families. Temple University Press. ISBN 978-1-59213-195-2.
- Serjeant, Graham R.; Beryl E. (2001). Sickle Cell Disease. Oxford University Press. ISBN 978-0-19-263036-0.
- Tapper, Melbourne (1999). In the blood: sickle cell anemia and the politics of race. University of Pennsylvania Press. ISBN 978-0-8122-3471-8.
External links
| Wikimedia Commons has media related to Sickle-cell disease. |
| Classification | |
|---|---|
| External resources |