Fanconi anemia
Fanconi anaemia (FA) is a rare genetic disease resulting in impaired response to DNA damage. Although it is a very rare disorder, study of this and other bone marrow failure syndromes has improved scientific understanding of the mechanisms of normal bone marrow function and development of cancer. Among those affected, the majority develop cancer, most often acute myelogenous leukemia, and 90% develop bone marrow failure (the inability to produce blood cells) by age 40. About 60–75% of people have congenital defects, commonly short stature, abnormalities of the skin, arms, head, eyes, kidneys, and ears, and developmental disabilities. Around 75% of people have some form of endocrine problems, with varying degrees of severity.
| Fanconi anaemia | |
|---|---|
| Other names | English: /fɑːnˈkoʊni/, /fæn-/ |
| Specialty | Hematology |
FA is the result of a genetic defect in a cluster of proteins responsible for DNA repair via homologous recombination.[1]
Treatment with androgens and hematopoietic (blood cell) growth factors can help bone marrow failure temporarily, but the long-term treatment is bone marrow transplant if a donor is available.[2] Because of the genetic defect in DNA repair, cells from people with FA are sensitive to drugs that treat cancer by DNA crosslinking, such as mitomycin C. The typical age of death was 30 years in 2000.[2]
FA occurs in about one per 130,000 births, with a higher frequency in Ashkenazi Jews and Afrikaners in South Africa.[3] The disease is named after the Swiss pediatrician who originally described this disorder, Guido Fanconi.[4][5] It should not be confused with Fanconi syndrome, a kidney disorder also named after Fanconi.
Signs and symptoms
FA is characterized by bone marrow failure, AML, solid tumors, and developmental abnormalities. Classic features include abnormal thumbs, absent radii, short stature, skin hyperpigmentation, including café au lait spots, abnormal facial features (triangular face, microcephaly), abnormal kidneys, and decreased fertility. Many FA patients (about 30%) do not have any of the classic physical findings, but Diepoxybutane chromosome fragility assay showing increased chromosomal breaks can make the diagnosis.[6] About 80% of FA will develop bone marrow failure by age 20.
The first sign of a hematologic problem is usually petechiae and bruises, with later onset of pale appearance, feeling tired, and infections. Because macrocytosis usually precedes a low platelet count, patients with typical congenital anomalies associated with FA should be evaluated for an elevated red blood cell mean corpuscular volume.[7]
Genetics
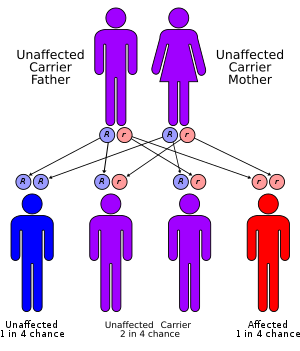
FA is primarily an autosomal recessive genetic disorder. This means that two mutated alleles (one from each parent) are required to cause the disease. The risk is 25% that each subsequent child will have FA. About 2% of FA cases are X-linked recessive, which means that if the mother carries one mutated Fanconi anemia allele on one X chromosome, a 50% chance exists that male offspring will present with Fanconi anemia.
Scientists have identified 22 FA or FA-like genes: FANCA, FANCB, FANCC, FANCD1 (BRCA2), FANCD2, FANCE, FANCF, FANCG, FANCI, FANCJ (BRIP1), FANCL, FANCM, FANCN (PALB2), FANCO (RAD51C), FANCP (SLX4), FANCQ (XPF), FANCS (BRCA1), FANCT (UBE2T), FANCU (XRCC2), FANCV (REV7), and FANCW (RFWD3). FANCB is the one exception to FA being autosomal recessive, as this gene is on the X chromosome. These genes are involved in DNA repair.
The carrier frequency in the Ashkenazi Jewish population is about one in 90.[8] Genetic counseling and genetic testing are recommended for families who may be carriers of Fanconi anemia.
Because of the failure of hematologic components—white blood cells, red blood cells, and platelets—to develop, the body's capabilities to fight infection, deliver oxygen, and form clots are all diminished.
Pathogenesis
Clinically, hematological abnormalities are the most serious symptoms in FA. By the age of 40, 98% of FA patients will have developed some type of hematological abnormality. However, a few cases have occurred in which older patients have died without ever developing them. Symptoms appear progressively, and often lead to complete bone marrow failure. While at birth, blood count is usually normal, macrocytosis/megaloblastic anemia, defined as unusually large red blood cells, is the first detected abnormality, often within the first decade of life (median age of onset is 7 years). Within the next 10 years, over 50% of patients presenting haematological abnormalities will have developed pancytopenia, defined as abnormalities in two or more blood cell lineages. This is in contrast to Diamond–Blackfan anemia, which affects only erythrocytes, and Shwachman–Diamond syndrome, which primarily causes neutropenia. Most commonly, a low platelet count (thrombocytopenia) precedes a low neutrophil count (neutropenia), with both appearing with relative equal frequencies. The deficiencies cause increased risk of hemorrhage and recurrent infections, respectively.
As FA is now known to affect DNA repair, specifically homologous recombination,[1] and given the current knowledge about dynamic cell division in the bone marrow, patients are consequently more likely to develop bone marrow failure, myelodysplastic syndromes, and acute myeloid leukemia (AML).
Myelodysplastic syndromes
MDSs, formerly known as preleukemia, are a group of bone marrow neoplastic diseases that share many of the morphologic features of AML, with some important differences. First, the percentage of undifferentiated progenitor cells, blast cells, is always less than 20%, with considerably more dysplasia, defined as cytoplasmic and nuclear morphologic changes in erythroid, granulocytic, and megakaryocytic precursors, than what is usually seen in cases of AML. These changes reflect delayed apoptosis or a failure of programmed cell death. When left untreated, MDS can lead to AML in about 30% of cases. Due the nature of the FA pathology, MDS diagnosis cannot be made solely through cytogenetic analysis of the marrow. Indeed, it is only when morphologic analysis of marrow cells is performed, that a diagnosis of MDS can be ascertained. Upon examination, MDS-afflicted FA patients will show many clonal variations, appearing either prior or subsequent to the MDS. Furthermore, cells will show chromosomal aberrations, the most frequent being monosomy 7 and partial trisomies of chromosome 3q 15. Observation of monosomy 7 within the marrow is well correlated with an increased risk of developing AML and with a very poor prognosis, death generally ensuing within 2 years (unless prompt allogeneic hematopoietic progenitor cell transplant is an option).[9]
Acute myeloid leukemia
FA patients are at elevated risk for the development of AML defined as presence of 20% or more of myeloid blasts in the marrow or 5 to 20% myeloid blasts in the blood. All of the subtypes of AML can occur in FA with the exception of promyelocytic. However, myelomonocytic and acute monocytic are the most common subtypes observed. Many MDS patients' diseases evolve into AML if they survive long enough. Furthermore, the risk of developing AML increases with the onset of bone-marrow failure.
Although risk of developing either MDS or AML before the age of 20 is only 27%, this risk increases to 43% by the age of 30 and 52% by the age of 40. Historically, even with a marrow transplant, about a quarter of FA patients diagnosed with MDS/ALS have died from MDS/ALS-related causes within two years,[10] although more recent published evidence suggests that earlier allogeneic hematopoietic progenitor cell transplantation in children with FA is leading to better outcomes over time.[11]
Bone marrow failure
The last major haematological complication associated with FA is bone marrow failure, defined as inadequate blood cell production. Several types of failure are observed in FA patients, and generally precede MDS and AML. Detection of decreasing blood count is generally the first sign used to assess necessity of treatment and possible transplant. While most FA patients are initially responsive to androgen therapy and haemopoietic growth factors, these have been shown to promote leukemia, especially in patients with clonal cytogenetic abnormalities, and have severe side effects, including hepatic adenomas and adenocarcinomas. The only treatment left would be bone marrow transplant; however, such an operation has a relatively low success rate in FA patients when the donor is unrelated (30% 5-year survival). It is, therefore, imperative to transplant from an HLA-identical sibling. Furthermore, due to the increased susceptibility of FA patients to chromosomal damage, pretransplant conditioning cannot include high doses of radiation or immunosuppressants, thus increased chances of patients developing graft-versus-host disease. If all precautions are taken, and the marrow transplant is performed within the first decade of life, two-year probability of survival can be as high as 89%. However, if the transplant is performed at ages older than 10, two-year survival rates drop to 54%.
A recent report by Zhang et al. investigates the mechanism of bone marrow failure in FANCC-/- cells.[12] They hypothesize and successfully demonstrate that continuous cycles of hypoxia-reoxygenation, such as those seen by haemopoietic and progenitor cells as they migrate between hyperoxic blood and hypoxic marrow tissues, leads to premature cellular senescence and therefore inhibition of haemopoietic function. Senescence, together with apoptosis, may constitute a major mechanism of haemopoietic cell depletion occurred in bone marrow failure.
Molecular basis
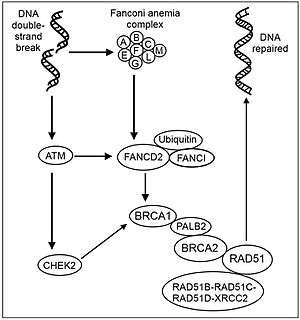
There are 19 genes responsible for FA, one of them being the breast-cancer susceptibility gene BRCA2. They are involved in the recognition and repair of damaged DNA; genetic defects leave them unable to repair DNA. The FA core complex of 8 proteins is normally activated when DNA stops replicating because of damage. The core complex adds ubiquitin, a small protein that combines with BRCA2 in another cluster to repair DNA (see Figure Recombinational repair of DNA double-strand damage). At the end of the process, ubiquitin is removed.[2]
Recent studies have shown that eight of these proteins, FANCA, -B, -C, -E, -F, -G, -L and -M, assemble to form a core protein complex in the nucleus. According to current models, the complex moves from the cytoplasm into the nucleus following nuclear localization signals on FANCA and FANCE. Assembly is activated by replicative stress, particularly DNA damage caused by cross-linking agents (such as mitomycin C or cisplatin) or reactive oxygen species (ROS) that is detected by the FANCM protein.[20]
Following assembly, the protein core complex activates FANCL protein which acts as an E3 ubiquitin-ligase and monoubiquitinates FANCD2.[21][22][23][24]
Monoubiquitinated FANCD2, also known as FANCD2-L, then goes on to interact with a BRCA1/BRCA2 complex (see Figure Recombinational repair of DNA double-strand damage). Details are not known, but similar complexes are involved in genome surveillance and associated with a variety of proteins implicated in DNA repair and chromosomal stability.[25][26] With a crippling mutation in any FA protein in the complex, DNA repair is much less effective, as shown by its response to damage caused by cross-linking agents such as cisplatin, diepoxybutane[27] and Mitomycin C. Bone marrow is particularly sensitive to this defect.
In another pathway responding to ionizing radiation, FANCD2 is thought to be phosphorylated by protein complex ATM/ATR activated by double-strand DNA breaks, and takes part in S-phase checkpoint control. This pathway was proven by the presence of radioresistant DNA synthesis, the hallmark of a defect in the S phase checkpoint, in patients with FA-D1 or FA-D2. Such a defect readily leads to uncontrollable replication of cells and might also explain the increase frequency of AML in these patients.
Spermatogenesis
In humans, infertility is one of the characteristics of individuals with mutational defects in the FANC genes.[28] In mice, spermatogonia, preleptotene spermatocytes, and spermatocytes in the meiotic stages of leptotene, zygotene and early pachytene are enriched for FANC proteins.[28] This finding suggests that recombinational repair processes mediated by the FANC proteins are active during germ cell development, particularly during meiosis, and that defects in this activity can lead to infertility.
Neural stem cell homeostasis
Microphthalmia and microcephaly are frequent congenital defects in FA patients. The loss of FANCA and FANCG in mice causes neural progenitor apoptosis both during early developmental neurogenesis and later during adult neurogenesis. This leads to depletion of the neural stem cell pool with aging.[29] Much of the Fanconi anemia phenotype might be interpreted as a reflection of premature aging of stem cells.[29]
Treatment
The first line of therapy is androgens and hematopoietic growth factors, but only 50–75% of patients respond. A more permanent cure is hematopoietic stem cell transplantation.[30] If no potential donors exist, a savior sibling can be conceived by preimplantation genetic diagnosis (PGD) to match the recipient's HLA type.[31][32]
Prognosis
Many patients eventually develop acute myelogenous leukemia (AML). Older patients are extremely likely to develop head and neck, esophageal, gastrointestinal, vulvar and anal cancers.[33] Patients who have had a successful bone marrow transplant and, thus, are cured of the blood problem associated with FA still must have regular examinations to watch for signs of cancer. Many patients do not reach adulthood.
The overarching medical challenge that Fanconi patients face is a failure of their bone marrow to produce blood cells. In addition, Fanconi patients normally are born with a variety of birth defects. A significant number of Fanconi patients have kidney problems, trouble with their eyes, developmental delay and other serious defects, such as microcephaly (small head).[34]
References
- Walden, Helen; Deans, Andrew J (April 17, 2014). "The Fanconi Anemia DNA Repair Pathway: Structural and Functional Insights into a Complex Disorder". Annu. Rev. Biophys. 43: 257–278. doi:10.1146/annurev-biophys-051013-022737. PMID 24773018.
- Schwartz, Robert S.; d'Andrea, Alan D. (May 2010). "Susceptibility pathways in Fanconi's anemia and breast cancer". N. Engl. J. Med. 362 (20): 1909–1919. doi:10.1056/NEJMra0809889. PMC 3069698. PMID 20484397.
- Rosenberg PS (2011). "How high are carrier frequencies of rare recessive syndromes? Contemporary estimates for Fanconi Anemia in the United States and Israel". American Journal of Medical Genetics Part A. 155 (8): 1877–1883. doi:10.1002/ajmg.a.34087. PMC 3140593. PMID 21739583.
- synd/61 at Who Named It?
- Fanconi, G. (1927). "Familiäre infantile perniziosaartige Anämie (perniziöses Blutbild und Konstitution)". Jahrbuch für Kinderheilkunde und physische Erziehung (Wien). 117: 257–280. (Commentary on and reprint ... doi:10.1016/B978-012448510-5.50106-0)
- Chirnomas SD, Kupfer GM (2013). "The inherited bone marrow failure syndromes". Pediatr Clin North Am. 60 (6): 1291–310. doi:10.1016/j.pcl.2013.09.007. PMC 3875142. PMID 24237972.
- Fanconi Anemia~clinical at eMedicine
- Kutler DI, Auerbach AD (2004). "Fanconi anemia in Ashkenazi Jews". Fam. Cancer. 3 (3–4): 241–248. doi:10.1007/s10689-004-9565-8. PMID 15516848.
- Mehta PA, et al. (2010). "Numerical chromosomal changes and risk of development of myelodysplastic syndrome-acute myeloid leukemia in patients with Fanconi anemia". Cancer Genetics. 203 (2): 180–186. doi:10.1016/j.cancergencyto.2010.07.127. PMID 21156231.
- Butturini A; et al. (1994). "Hematologic abnormalities in Fanconi anemia: an International Fanconi Anemia Registry study". Blood. 84 (5): 1650–4. doi:10.1182/blood.v84.5.1650.bloodjournal8451650. PMID 8068955.
- de Latour RP, et al. (2013). "Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the EBMT experience". Blood. 122 (26): 4279–4286. doi:10.1182/blood-2013-01-479733. PMID 24144640.
- Zhang X, Li J, Sejas DP, Pang Q (2005). "Hypoxia-reoxygenation induces premature senescence in FA bone marrow hematopoietic cells". Blood. 106 (1): 75–85. doi:10.1182/blood-2004-08-3033. PMID 15769896. S2CID 14059745.
- D'Andrea AD (2010). "Susceptibility pathways in Fanconi's anemia and breast cancer". N. Engl. J. Med. 362 (20): 1909–19. doi:10.1056/NEJMra0809889. PMC 3069698. PMID 20484397.
- Sobeck A, Stone S, Landais I, de Graaf B, Hoatlin ME (2009). "The Fanconi anemia protein FANCM is controlled by FANCD2 and the ATR/ATM pathways". J. Biol. Chem. 284 (38): 25560–8. doi:10.1074/jbc.M109.007690. PMC 2757957. PMID 19633289.
- Castillo P, Bogliolo M, Surralles J (2011). "Coordinated action of the Fanconi anemia and ataxia telangiectasia pathways in response to oxidative damage". DNA Repair (Amst.). 10 (5): 518–25. doi:10.1016/j.dnarep.2011.02.007. PMID 21466974.
- Stolz A, Ertych N, Bastians H (2011). "Tumor suppressor CHK2: regulator of DNA damage response and mediator of chromosomal stability". Clin. Cancer Res. 17 (3): 401–5. doi:10.1158/1078-0432.CCR-10-1215. PMID 21088254.
- Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D'Andrea AD (2002). "S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51". Blood. 100 (7): 2414–20. doi:10.1182/blood-2002-01-0278. PMID 12239151.
- Park JY, Zhang F, Andreassen PR (2014). "PALB2: the hub of a network of tumor suppressors involved in DNA damage responses". Biochim. Biophys. Acta. 1846 (1): 263–75. doi:10.1016/j.bbcan.2014.06.003. PMC 4183126. PMID 24998779.
- Chun J, Buechelmaier ES, Powell SN (2013). "Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway". Mol. Cell. Biol. 33 (2): 387–95. doi:10.1128/MCB.00465-12. PMC 3554112. PMID 23149936.
- Deans AJ, West SC (December 2009). "FANCM connects the genome instability disorders Bloom's Syndrome and Fanconi Anemia". Mol. Cell. 36 (6): 943–53. doi:10.1016/j.molcel.2009.12.006. PMID 20064461.
- Vandenberg CJ; Gergely F; Ong CY; et al. (2003). "BRCA1-independent ubiquitination of FANCD2". Mol. Cell. 12 (1): 247–254. doi:10.1016/S1097-2765(03)00281-8. PMID 12887909.
- Garcia-Higuera I; Taniguchi T; Ganesan S; et al. (2001). "Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway". Mol. Cell. 7 (2): 249–262. doi:10.1016/S1097-2765(01)00173-3. PMID 11239454.
- Wang Y, Cortez D, Yazdi P, Neff N, Elledge SJ, Qin J (2000). "BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures". Genes Dev. 14 (8): 927–39. doi:10.1101/gad.14.8.927 (inactive 2020-06-07). PMC 316544. PMID 10783165.
- Cortez D, Wang Y, Qin J, Elledge SJ (1999). "Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks". Science. 286 (5442): 1162–1166. doi:10.1126/science.286.5442.1162. PMID 10550055.
- Howlett NG; Taniguchi T; Olson S; et al. (2002). "Biallelic inactivation of BRCA2 in Fanconi anemia". Science. 297 (5581): 606–609. Bibcode:2002Sci...297..606H. doi:10.1126/science.1073834. PMID 12065746.
- Connor F; Bertwistle D; Mee PJ; et al. (1997). "Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation". Nat. Genet. 17 (4): 423–430. doi:10.1038/ng1297-423. PMID 9398843.
- Auerbach AD, Rogatko A, Schroeder-Kurth TM (1989). "International Fanconi Anemia Registry: relation of clinical symptoms to diepoxybutane sensitivity". Blood. 73 (2): 391–6. doi:10.1007/978-3-642-74179-1_1. ISBN 978-3-642-74181-4. PMID 2917181.
- Jamsai D, O'Connor AE, O'Donnell L, Lo JC, O'Bryan MK (2015). "Uncoupling of transcription and translation of Fanconi anemia (FANC) complex proteins during spermatogenesis". Spermatogenesis. 5 (1): e979061. doi:10.4161/21565562.2014.979061. PMC 4581071. PMID 26413409.
- Sii-Felice K, Barroca V, Etienne O, Riou L, Hoffschir F, Fouchet P, Boussin FD, Mouthon MA (2008). "Role of Fanconi DNA repair pathway in neural stem cell homeostasis". Cell Cycle. 7 (13): 1911–5. doi:10.4161/cc.7.13.6235. PMID 18604174.
- Fanconi Anemia~treatment at eMedicine
- Page 29 in Moore, Pete (2007). The Debate About Genetic Engineering (Ethical Debates). New York, NY: Rosen Central. ISBN 978-1-4042-3754-4.
- Verlinsky, Y; Rechitsky, S; Schoolcraft, W; Strom, C; Kuliev, A (2001). "Preimplantation diagnosis for Fanconi anemia combined with HLA matching". JAMA. 285 (24): 3130–3133. doi:10.1001/jama.285.24.3130. PMID 11427142.
- Institut Biologia Fonamental de Barcelona, "Constitutional chromosomal instability: a case with three primary and sequential cancers" Archived 2010-10-03 at the Wayback Machine, USUJ 2009. Retrieved 2010-04-13
- Anemia, Fanconi Archived February 22, 2014, at the Wayback Machine
External links
| Classification | |
|---|---|
| External resources |
- Fanconi Anemia Research Fund
- GeneReviews/NCBI/NIH/UW entry on Fanconi Anemia
- OMIM entries on Fanconi Anemia
- Fanconi anemia at Curlie
- Fanconi Hope Charitable Trust - based in the UK, with Eu and International Links
- Fanconi Anaemia FAmily Support - based in the UK
- Fanconi's Anaemia on patient.info