Geochemistry
Geochemistry is the science that uses the tools and principles of chemistry to explain the mechanisms behind major geological systems such as the Earth's crust and its oceans.[1]:1 The realm of geochemistry extends beyond the Earth, encompassing the entire Solar System,[2] and has made important contributions to the understanding of a number of processes including mantle convection, the formation of planets and the origins of granite and basalt.[1]:1 It is an integrated field of chemistry and geology/geography.
History

The term geochemistry was first used by the Swiss-German chemist Christian Friedrich Schönbein in 1838: "a comparative geochemistry ought to be launched, before geochemistry can become geology, and before the mystery of the genesis of our planets and their inorganic matter may be revealed."[3] However, for the rest of the century the more common term was "chemical geology", and there was little contact between geologists and chemists.[3]
Geochemistry emerged as a separate discipline after major laboratories were established, starting with the United States Geological Survey (USGS) in 1884, and began systematic surveys of the chemistry of rocks and minerals. The chief USGS chemist, Frank Wigglesworth Clarke, noted that the elements generally decrease in abundance as their atomic weights increase, and summarized the work on elemental abundance in The Data of Geochemistry.[3][4]:2
The composition of meteorites was investigated and compared to terrestrial rocks as early as 1850. In 1901, Oliver C. Farrington hypothesised that, although there were differences, the relative abundances should still be the same.[3] This was the beginnings of the field of cosmochemistry and has contributed much of what we know about the formation of the Earth and the Solar System.[5]
In the early 20th century, Max von Laue and William L. Bragg showed that X-ray scattering could be used to determine the structures of crystals. In the 1920s and 1930s, Victor Goldschmidt and associates at the University of Oslo applied these methods to many common minerals and formulated a set of rules for how elements are grouped. Goldschmidt published this work in the series Geochemische Verteilungsgesetze der Elemente [Geochemical Laws of the Distribution of Elements].[4]:2[6]
Subfields
Some subfields of geochemistry are:[7]
- Aqueous geochemistry studies the role of various elements in watersheds, including copper, sulfur, mercury, and how elemental fluxes are exchanged through atmospheric-terrestrial-aquatic interactions.[8]
- Biogeochemistry is the field of study focusing on the effect of life on the chemistry of the Earth.[9]:3
- Cosmochemistry includes the analysis of the distribution of elements and their isotopes in the cosmos.[2]:1
- Isotope geochemistry involves the determination of the relative and absolute concentrations of the elements and their isotopes in the Earth and on Earth's surface.[10]
- Organic geochemistry, the study of the role of processes and compounds that are derived from living or once-living organisms.[11]
- Photogeochemistry is the study of light-induced chemical reactions that occur or may occur among natural components of the Earth's surface.[12]
- Regional geochemistry includes applications to environmental, hydrological and mineral exploration studies.[13]
Chemical elements
The building blocks of materials are the chemical elements. These can be identified by their atomic number Z, which is the number of protons in the nucleus. An element can have more than one value for N, the number of neutrons in the nucleus. The sum of these is the mass number, which is roughly equal to the atomic mass. Atoms with the same atomic number but different neutron numbers are called isotopes. A given isotope is identified by a letter for the element preceded by a superscript for the mass number. For example, two common isotopes of chlorine are 35Cl and 37Cl. There are about 1700 known combinations of Z and N, of which only about 260 are stable. However, most of the unstable isotopes do not occur in nature. In geochemistry, stable isotopes are used to trace chemical pathways and reactions, while isotopes are primarily used to date samples.[4]:13–17
The chemical behavior of an atom – its affinity for other elements and the type of bonds it forms – is determined by the arrangement of electrons in orbitals, particularly the outermost (valence) electrons. These arrangements are reflected in the position of elements in the periodic table.[4]:13–17 Based on position, the elements fall into the broad groups of alkali metals, alkaline earth metals, transition metals, semi-metals (also known as metalloids), halogens, noble gases, lanthanides and actinides.[4]:20–23
Another useful classification scheme for geochemistry is the Goldschmidt classification, which places the elements into four main groups. Lithophiles combine easily with oxygen. These elements, which include Na, K, Si, Al, Ti, Mg and Ca, dominate in the Earth's crust, forming silicates and other oxides. Siderophile elements (Fe, Co, Ni, Pt, Re, Os) have an affinity for iron and tend to concentrate in the core. Chalcophile elements (Cu, Ag, Zn, Pb, S) form sulfides; and atmophile elements (O, N, H and noble gases) dominate the atmosphere. Within each group, some elements are refractory, remaining stable at high temperatures, while others are volatile, evaporating more easily, so heating can separate them.[1]:17[4]:23
Differentiation and mixing
The chemical composition of the Earth and other bodies is determined by two opposing processes: differentiation and mixing. In the Earth's mantle, differentiation occurs at mid-ocean ridges through partial melting, with more refractory materials remaining at the base of the lithosphere while the remainder rises to form basalt. After an oceanic plate descends into the mantle, convection eventually mixes the two parts together. Erosion differentiates granite, separating it into clay on the ocean floor, sandstone on the edge of the continent, and dissolved minerals in ocean waters. Metamorphism and anatexis (partial melting of crustal rocks) can mix these elements together again. In the ocean, biological organisms can cause chemical differentiation, while dissolution of the organisms and their wastes can mix the materials again.[1]:23–24
Fractionation
A major source of differentiation is fractionation, an unequal distribution of elements and isotopes. This can be the result of chemical reactions, phase changes, kinetic effects, or radioactivity.[1]:2–3 On the largest scale, planetary differentiation is a physical and chemical separation of a planet into chemically distinct regions. For example, the terrestrial planets formed iron-rich cores and silicate-rich mantles and crusts.[14]:218 In the Earth's mantle, the primary source of chemical differentiation is partial melting, particularly near mid-ocean ridges.[15]:68,153 This can occur when the solid is heterogeneous or a solid solution, and part of the melt is separated from the solid. The process is known as equilibrium or batch melting if the solid and melt remain in equilibrium until the moment that the melt is removed, and fractional or Rayleigh melting if it is removed continuously.[16]
Isotopic fractionation can have mass-dependent and mass-independent forms. Molecules with heavier isotopes have lower ground state energies and are therefore more stable. As a result, chemical reactions show a small isotope dependence, with heavier isotopes preferring species or compounds with a higher oxidation state; and in phase changes, heavier isotopes tend to concentrate in the heavier phases.[17] Mass-dependent fractionation is largest in light elements because the difference in masses is a larger fraction of the total mass.[18]:47
Ratios between isotopes are generally compared to a standard. For example, sulfur has four stable isotopes, of which the two most common are 32S and 34S.[18]:98 The ratio of their concentrations, R=34S/32S, is reported as

where Rs is the same ratio for a standard. Because the differences are small, the ratio is multiplied by 1000 to make it parts per thousand (referred to as parts per mil). This is represented by the symbol ‰.[17]:55
Equilibrium
Equilibrium fractionation occurs between chemicals or phases that are in equilibrium with each other. In equilibrium fractionation between phases, heavier phases prefer the heavier isotopes. For two phases A and B, the effect can be represented by the factor

In the liquid-vapor phase transition for water, al-v at 20 degrees Celsius is 1.0098 for 18O and 1.084 for 2H. In general, fractionation is greater at lower temperatures. At 0 °C, the factors are 1.0117 and 1.111.[17]:59
Kinetic
When there is not equilibrium between phases or chemical compounds, kinetic fractionation can occur. For example, at interfaces between liquid water and air, the forward reaction is enhanced if the humidity of the air is less than 100% or the water vapor is moved by a wind. Kinetic fractionation generally is enhanced compared to equilibrium fractionation, and depends on factors such as reaction rate, reaction pathway and bond energy. Since lighter isotopes generally have weaker bonds, they tend to react faster and enrich the reaction products.[17]:60
Biological fractionation is a form of kinetic fractionation, since reactions tend to be in one direction. Biological organisms prefer lighter isotopes because there is a lower energy cost in breaking energy bonds. In addition to the previously mentioned factors, the environment and species of the organism can have a large effect on the fractionation.[17]:70
Cycles
Through a variety of physical and chemical processes, chemical elements change in concentration and move around in what are called geochemical cycles. An understanding of these changes requires both detailed observation and theoretical models. Each chemical compound, element or isotope has a concentration that is a function C(r,t) of position and time, but it is impractical to model the full variability. Instead, in an approach borrowed from chemical engineering,[1]:81 geochemists average the concentration over regions of the Earth called geochemical reservoirs. The choice of reservoir depends on the problem; for example, the ocean may be a single reservoir or be split into multiple reservoirs.[19] In a type of model called a box model, a reservoir is represented by a box with inputs and outputs.[1]:81[19]
Geochemical models generally involve feedback. In the simplest case of a linear cycle, either the input or the output from a reservoir is proportional to the concentration. For example, salt is removed from the ocean by formation of evaporites, and given a constant rate of evaporation in evaporite basins, the rate of removal of salt should be proportional to its concentration. For a given component C, if the input to a reservoir is a constant a and the output is kC for some constant k, then the mass balance equation is
-
(1)

This expresses the fact that any change in mass must be balanced by changes in the input or output. On a time scale of t = 1/k, the system approaches a steady state in which Csteady = a/k. The residence time is defined as

where I and O are the input and output rates. In the above example, the steady-state input and output rates are both equal to a, so τres = 1/k.[19]
If the input and output rates are nonlinear functions of C, they may still be closely balanced over time scales much greater than the residence time; otherwise there will be large fluctuations in C. In that case, the system is always close to a steady state and a lowest order expansion of the mass balance equation will lead to a linear equation like Equation (1). In most systems, one or both of the input and output depend on C, resulting in a feedback that tends to maintain the steady state. If an external forcing perturbs the system, it will return to the steady state on a time scale of 1/k.[19]
Abundance of elements
Solar System
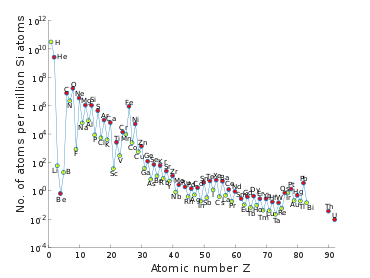
The composition of the Solar System is similar to that of many other stars, and aside from small anomalies it can be assumed to have formed from a solar nebula that had a uniform composition, and the composition of the Sun's photosphere is similar to that of the rest of the Solar System. The composition of the photosphere is determined by fitting the absorption lines in its spectrum to models of the Sun's atmosphere.[21] By far the largest two elements by fraction of total mass are hydrogen (74.9%) and helium (23.8%), with all the remaining elements contributing just 1.3%.[22] There is a general trend of exponential decrease in abundance with increasing atomic number, although elements with even atomic number are more common than their odd-numbered neighbors (the Oddo–Harkins rule). Compared to the overall trend, lithium, boron and beryllium are depleted and iron is anomalously enriched.[23]:284–285
The pattern of elemental abundance is mainly due to two factors. The hydrogen, helium, and some of the lithium were formed in about 20 minutes after the Big Bang, while the rest were created in the interiors of stars.[4]:316–317
Meteorites
Meteorites come in a variety of compositions, but chemical analysis can determine whether they were once in planetesimals that melted or differentiated.[21]:45 Chondrites are undifferentiated and have round mineral inclusions called chondrules. With ages of 4.56 billion years, they date to the early solar system. A particular kind, the CI chondrite, has a composition that closely matches that of the Sun's photosphere, except for depletion of some volatiles (H, He, C, N, O) and a group of elements (Li, B, Be) that are destroyed by nucleosynthesis in the Sun.[4]:318[21] Because of the latter group, CI chondrites are considered a better match for the composition of the early Solar System. Moreover, the chemical analysis of CI chondrites is more accurate than for the photosphere, so it is generally used as the source for chemical abundance, despite their rareness (only five have been recovered on Earth).[21]
Giant planets
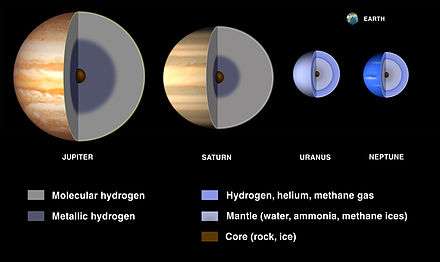
The planets of the Solar System are divided into two groups: the four inner planets are the terrestrial planets (Mercury, Venus, Earth and Mars), with relatively small sizes and rocky surfaces. The four outer planets are the giant planets, which are dominated by hydrogen and helium and have lower mean densities. These can be further subdivided into the gas giants (Jupiter and Saturn) and the ice giants (Uranus and Neptune) that have large icy cores.[24]:26–27,283–284
Most of our direct information on the composition of the giant planets is from spectroscopy. Since the 1930s, Jupiter was known to contain hydrogen, methane and ammonium. In the 1960s, interferometry greatly increased the resolution and sensitivity of spectral analysis, allowing the identification of a much greater collection of molecules including ethane, acetylene, water and carbon monoxide.[25]:138–139 However, Earth-based spectroscopy becomes increasingly difficult with more remote planets, since the reflected light of the Sun is much dimmer; and spectroscopic analysis of light from the planets can only be used to detect vibrations of molecules, which are in the infrared frequency range. This constrains the abundances of the elements H, C and N.[25]:130 Two other elements are detected: phosphorus in the gas phosphine (PH3) and germanium in germane (GeH4).[25]:131
The helium atom has vibrations in the ultraviolet range, which is strongly absorbed by the atmospheres of the outer planets and Earth. Thus, despite its abundance, helium was only detected once spacecraft were sent to the outer planets, and then only indirectly through collision-induced absorption in hydrogen molecules.[25]:209 Further information on Jupiter was obtained from the Galileo probe when it was sent into the atmosphere in 1995;[26][27] and the final mission of the Cassini probe in 2017 was to enter the atmosphere of Saturn.[28] In the atmosphere of Jupiter, He was found to be depleted by a factor of 2 compared to solar composition and Ne by a factor of 10, a surprising result since the other noble gases and the elements C, N and S were enhanced by factors of 2 to 4 (oxygen was also depleted but this was attributed to the unusually dry region that Galileo sampled).[27]
Spectroscopic methods only penetrate the atmospheres of Jupiter and Saturn to depths where the pressure is about equal to 1 bar, approximately Earth's atmospheric pressure at sea level.[25]:131 The Galileo probe penetrated to 22 bars.[27] This is a small fraction of the planet, which is expected to reach pressures of over 40 Mbar. To constrain the composition in the interior, thermodynamic models are constructed using information on temperature from infrared emission spectra and equations of state for the likely compositions.[25]:131 High pressure experiments predict that hydrogen will be a metallic liquid in the interior of Jupiter and Saturn, while in Uranus and Neptune it remains in the molecular state.[25]:135–136 Estimates also depend on models for the formation of the planets. Condensation of the presolar nebula would result in a gaseous planet with the same composition as the Sun, but the planets could also have formed when a solid core captured nebular gas.[25]:136
In current models, the four giant planets have cores of rock and ice that are roughly the same size, but the proportion of hydrogen and helium decreases from about 300 Earth masses in Jupiter to 75 in Saturn and just a few in Uranus and Neptune.[25]:220 Thus, while the gas giants are primarily composed of hydrogen and helium, the ice giants are primarily composed of heavier elements (O, C, N, S), primarily in the form of water, methane and ammonia. The surfaces are cold enough for molecular hydrogen to be liquid, so much of each planet is likely a hydrogen ocean overlaying one of heavier compounds.[29] Outside the core, Jupiter has a mantle of liquid metallic hydrogen and an atmosphere of molecular hydrogen and helium. Metallic hydrogen does not mix well with helium, and in Saturn it may form a separate layer below the metallic hydrogen.[25]:138
Terrestrial planets
Terrestrial planets are believed to have come from the same nebular material as the giant planets, but they have lost most of the lighter elements and have different histories. Planets closer to the Sun might be expected to have a higher fraction of refractory elements, but if their later stages of formation involved collisions of large objects with orbits that sampled different parts of the Solar System, there could be little systematic dependence on position.[30]:3–4
Direct information on Mars, Venus and Mercury largely comes from spacecraft missions. Using gamma-ray spectrometers, the composition of the crust of Mars has been measured by the Mars Odyssey orbiter,[31] the crust of Venus by some of the Venera missions to Venus,[30] and the crust of Mercury by the MESSENGER spacecraft.[32] Additional information on Mars comes from meteorites that have landed on Earth (the Shergottites, Nakhlites, and Chassignites, collectively known as SNC meteorites).[33]:124 Abundances are also constrained by the masses of the planets, while the internal distribution of elements is constrained by their moments of inertia.[4]:334
The planets condensed from the solar nebula, and much of the details of their composition are determined by fractionation as they cooled. The phases that condense fall into five groups. First to condense are materials rich in refractory elements such as Ca and Al. These are followed by nickel and iron, then magnesium silicates. Below about 700 kelvins (700 K), FeS and volatile-rich metals and silicates form a fourth group, and in the fifth group FeO enter the magnesium silicates.[34] The compositions of the planets and the Moon are chondritic, meaning that within each group the ratios between elements are the same as in carbonaceous chondrites.[4]:334
The estimates of planetary compositions depend on the model used. In the equilibrium condensation model, each planet was formed from a feeding zone in which the compositions of solids were determined by the temperature in that zone. Thus, Mercury formed at 1400 K, where iron remained in a pure metallic form and there was little magnesium or silicon in solid form; Venus at 900 K, so all the magnesium and silicon condensed; Earth at 600 K, so it contains FeS and silicates; and Mars at 450 K, so FeO was incorporated into magnesium silicates. The greatest problem with this theory is that volatiles would not condense, so the planets would have no atmospheres and Earth no atmosphere.[4]:335–336
In chondritic mixing models, the compositions of chondrites are used to estimate planetary compositions. For example, one model mixes two components, one with the composition of C1 chondrites and one with just the refractory components of C1 chondrites.[4]:337 In another model, the abundances of the five fractionation groups are estimated using an index element for each group. For the most refractory group, uranium is used; iron for the second; the ratios of potassium and thallium to uranium for the next two; and the molar ratio FeO/(FeO+MgO) for the last. Using thermal and seismic models along with heat flow and density, Fe can be constrained to within 10 percent on Earth, Venus and Mercury. U can be constrained within about 30% on Earth, but its abundance on other planets is based on "educated guesses". One difficulty with this model is that there may be significant errors in its prediction of volatile abundances because some volatiles are only partially condensed.[34][4]:337–338
Earth's crust
The more common rock constituents are nearly all oxides; chlorides, sulfides and fluorides are the only important exceptions to this and their total amount in any rock is usually much less than 1%. By 1911, F. W. Clarke had calculated that a little more than 47% of the Earth's crust consists of oxygen. It occurs principally in combination as oxides, of which the chief are silica, alumina, iron oxides, and various carbonates (calcium carbonate, magnesium carbonate, sodium carbonate, and potassium carbonate). The silica functions principally as an acid, forming silicates, and all the commonest minerals of igneous rocks are of this nature. From a computation based on 1672 analyses of numerous kinds of rocks Clarke arrived at the following as the average percentage composition of the Earth's crust: SiO2=59.71, Al2O3=15.41, Fe2O3=2.63, FeO=3.52, MgO=4.36, CaO=4.90, Na2O=3.55, K2O=2.80, H2O=1.52, TiO2=0.60, P2O5=0.22, (total 99.22%). All the other constituents occur only in very small quantities, usually much less than 1%.[35]
These oxides combine in a haphazard way. For example, potash (potassium carbonate) and soda (sodium carbonate) combine to produce feldspars. In some cases they may take other forms, such as nepheline, leucite, and muscovite, but in the great majority of instances they are found as feldspar. Phosphoric acid with lime (calcium carbonate) forms apatite. Titanium dioxide with ferrous oxide gives rise to ilmenite. Part of the lime forms lime feldspar. Magnesium carbonate and iron oxides with silica crystallize as olivine or enstatite, or with alumina and lime form the complex ferro-magnesian silicates of which the pyroxenes, amphiboles, and biotites are the chief. Any excess of silica above what is required to neutralize the bases will separate out as quartz; excess of alumina crystallizes as corundum. These must be regarded only as general tendencies. It is possible, by rock analysis, to say approximately what minerals the rock contains, but there are numerous exceptions to any rule.[35]
Mineral constitution
Except in acid or siliceous igneous rocks containing greater than 66% of silica, known as felsic rocks, quartz is not abundant in igneous rocks. In basic rocks (containing 20% of silica or less) it is rare for them to contain as much silicon, these are referred to as mafic rocks. If magnesium and iron are above average while silica is low, olivine may be expected; where silica is present in greater quantity over ferro-magnesian minerals, such as augite, hornblende, enstatite or biotite, occur rather than olivine. Unless potash is high and silica relatively low, leucite will not be present, for leucite does not occur with free quartz. Nepheline, likewise, is usually found in rocks with much soda and comparatively little silica. With high alkalis, soda-bearing pyroxenes and amphiboles may be present. The lower the percentage of silica and alkali's, the greater is the prevalence of plagioclase feldspar as contracted with soda or potash feldspar.[35]
Earth's crust is composed of 90% silicate minerals and their abundance in the Earth is as follows: plagioclase feldspar (39%), alkali feldspar (12%), quartz (12%), pyroxene (11%), amphiboles (5%), micas (5%), clay minerals (5%); the remaining silicate minerals make up another 3% of Earth's crust. Only 8% of the Earth is composed of non-silicate minerals such as carbonates, oxides, and sulfides.[36]
The other determining factor, namely the physical conditions attending consolidation, plays on the whole a smaller part, yet is by no means negligible. Certain minerals are practically confined to deep-seated intrusive rocks, e.g., microcline, muscovite, diallage. Leucite is very rare in plutonic masses; many minerals have special peculiarities in microscopic character according to whether they crystallized in depth or near the surface, e.g., hypersthene, orthoclase, quartz. There are some curious instances of rocks having the same chemical composition, but consisting of entirely different minerals, e.g., the hornblendite of Gran, in Norway, which contains only hornblende, has the same composition as some of the camptonites of the same locality that contain feldspar and hornblende of a different variety. In this connection we may repeat what has been said above about the corrosion of porphyritic minerals in igneous rocks. In rhyolites and trachytes, early crystals of hornblende and biotite may be found in great numbers partially converted into augite and magnetite. Hornblende and biotite were stable under the pressures and other conditions below the surface, but unstable at higher levels. In the ground-mass of these rocks, augite is almost universally present. But the plutonic representatives of the same magma, granite and syenite contain biotite and hornblende far more commonly than augite.[35]
Felsic, intermediate and mafic igneous rocks
Those rocks that contain the most silica, and on crystallizing yield free quartz, form a group generally designated the "felsic" rocks. Those again that contain least silica and most magnesia and iron, so that quartz is absent while olivine is usually abundant, form the "mafic" group. The "intermediate" rocks include those characterized by the general absence of both quartz and olivine. An important subdivision of these contains a very high percentage of alkalis, especially soda, and consequently has minerals such as nepheline and leucite not common in other rocks. It is often separated from the others as the "alkali" or "soda" rocks, and there is a corresponding series of mafic rocks. Lastly a small sub-group rich in olivine and without feldspar has been called the "ultramafic" rocks. They have very low percentages of silica but much iron and magnesia.
Except these last, practically all rocks contain felspars or feldspathoid minerals. In the acid rocks the common feldspars are orthoclase, perthite, microcline, and oligoclase—all having much silica and alkalis. In the mafic rocks labradorite, anorthite and bytownite prevail, being rich in lime and poor in silica, potash and soda. Augite is the most common ferro-magnesian in mafic rocks, but biotite and hornblende are on the whole more frequent in felsic rocks.[35]
| Most Common Minerals | Felsic | Intermediate | Mafic | Ultramafic | |
|---|---|---|---|---|---|
| Quartz Orthoclase (and Oligoclase), Mica, Hornblende, Augite |
Little or no Quartz: Orthoclase hornblende, Augite, Biotite |
Little or no Quartz: Plagioclase Hornblende, Augite, Biotite |
No Quartz Plagioclase Augite, Olivine |
No Felspar Augite, Hornblende, Olivine | |
| Plutonic or Abyssal type | Granite | Syenite | Diorite | Gabbro | Peridotite |
| Intrusive or Hypabyssal type | Quartz-porphyry | Orthoclase-porphyry | Porphyrite | Dolerite | Picrite |
| Lavas or Effusive type | Rhyolite, Obsidian | Trachyte | Andesite | Basalt | Komatiite |
Rocks that contain leucite or nepheline, either partly or a wholly replacing felspar, are not included in this table. They are essentially of intermediate or of mafic character. We might in consequence regard them as varieties of syenite, diorite, gabbro, etc., in which feldspathoid minerals occur, and indeed there are many transitions between syenites of ordinary type and nepheline — or leucite — syenite, and between gabbro or dolerite and theralite or essexite. But, as many minerals develop in these "alkali" rocks that are uncommon elsewhere, it is convenient in a purely formal classification like that outlined here to treat the whole assemblage as a distinct series.[35]
| Most Common Minerals | Alkali Feldspar, Nepheline or Leucite, Augite, Hornblend, Biotite | Soda Lime Feldspar, Nepheline or Leucite, Augite, Hornblende (Olivine) | Nepheline or Leucite, Augite, Hornblende, Olivine |
|---|---|---|---|
| Plutonic type | Nepheline-syenite, Leucite-syenite, Nepheline-porphyry | Essexite and Theralite | Ijolite and Missourite |
| Effusive type or Lavas | Phonolite, Leucitophyre | Tephrite and Basanite | Nepheline-basalt, Leucite-basalt |
This classification is based essentially on the mineralogical constitution of the igneous rocks. Any chemical distinctions between the different groups, though implied, are relegated to a subordinate position. It is admittedly artificial but it has grown up with the growth of the science and is still adopted as the basis on which more minute subdivisions are erected. The subdivisions are by no means of equal value. The syenites, for example, and the peridotites, are far less important than the granites, diorites and gabbros. Moreover, the effusive andesites do not always correspond to the plutonic diorites but partly also to the gabbros. As the different kinds of rock, regarded as aggregates of minerals, pass gradually into one another, transitional types are very common and are often so important as to receive special names. The quartz-syenites and nordmarkites may be interposed between granite and syenite, the tonalites and adamellites between granite and diorite, the monzoaites between syenite and diorite, norites and hyperites between diorite and gabbro, and so on.[35]
Trace metals in the ocean
Trace metals readily form complexes with major ions in the ocean, including hydroxide, carbonate, and chloride and their chemical speciation changes depending on whether the environment is oxidized or reduced.[37] Benjamin (2002) defines complexes of metals with more than one type of ligand, other than water, as mixed-ligand-complexes. In some cases, a ligand contains more than one donor atom, forming very strong complexes, also called chelates (the ligand is the chelator). One of the most common chelators is EDTA (ethylenediaminetetraacetic acid), which can replace six molecules of water and form strong bonds with metals that have a plus two charge.[38] With stronger complexation, lower activity of the free metal ion is observed. One consequence of the lower reactivity of complexed metals compared to the same concentration of free metal is that the chelation tends to stabilize metals in the aqueous solution instead of in solids.[38]
Concentrations of the trace metals cadmium, copper, molybdenum, manganese, rhenium, uranium and vanadium in sediments record the redox history of the oceans. Within aquatic environments, cadmium(II) can either be in the form CdCl+(aq) in oxic waters or CdS(s) in a reduced environment. Thus higher concentrations of Cd in marine sediments may indicate low redox potential conditions in the past. For copper(II), a prevalent form is CuCl+(aq) within oxic environments and CuS(s) and Cu2S within reduced environments. The reduced seawater environment leads to two possible oxidation states of copper, Cu(I) and Cu(II). Molybdenum is present as the Mo(VI) oxidation state as MoO42−(aq) in oxic environments. Mo(V) and Mo(IV) are present in reduced environments in the forms MoO2+(aq) and MoS2(s). Rhenium is present as the Re(VII) oxidation state as ReO4− within oxic conditions, but is reduced to Re(IV) which may form ReO2 or ReS2. Uranium is in oxidation state VI in UO2(CO3)34−(aq) and is found in the reduced form UO2(s). Vanadium is in several forms in oxidation state V(V); HVO42− and H2VO4−. Its reduced forms can include VO2+, VO(OH)3−, and V(OH)3. These relative dominance of these species depends on pH.
In the water column of the ocean or deep lakes, vertical profiles of dissolved trace metals are characterized as following conservative–type, nutrient–type, or scavenged–type distributions. Across these three distributions, trace metals have different residence times and are used to varying extents by planktonic microorganisms. Trace metals with conservative-type distributions have high concentrations relative to their biological use. One example of a trace metal with a conservative-type distribution is molybdenum. It has a residence time within the oceans of around 8 x 105 years and is generally present as the molybdate anion (MoO42−). Molybdenum interacts weakly with particles and displays an almost uniform vertical profile in the ocean. Relative to the abundance of molybdenum in the ocean, the amount required as a metal cofactor for enzymes in marine phytoplankton is negligible.[39]
Trace metals with nutrient-type distributions are strongly associated with the internal cycles of particulate organic matter, especially the assimilation by plankton. The lowest dissolved concentrations of these metals are at the surface of the ocean, where they are assimilated by plankton. As dissolution and decomposition occur at greater depths, concentrations of these trace metals increase. Residence times of these metals, such as zinc, are several thousand to one hundred thousand years. Finally, an example of a scavenged-type trace metal is aluminium, which has strong interactions with particles as well as a short residence time in the ocean. The residence times of scavenged-type trace metals are around 100 to 1000 years. The concentrations of these metals are highest around bottom sediments, hydrothermal vents, and rivers. For aluminium, atmospheric dust provides the greatest source of external inputs into the ocean.[39]
Iron and copper show hybrid distributions in the ocean. They are influenced by recycling and intense scavenging. Iron is a limiting nutrient in vast areas of the oceans, and is found in high abundance along with manganese near hydrothermal vents. Here, many iron precipitates are found, mostly in the forms of iron sulfides and oxidized iron oxyhydroxide compounds. Concentrations of iron near hydrothermal vents can be up to one million times the concentrations found in the open ocean.[39]
Using electrochemical techniques, it is possible to show that bioactive trace metals (zinc, cobalt, cadmium, iron and copper) are bound by organic ligands in surface seawater. These ligand complexes serve to lower the bioavailability of trace metals within the ocean. For example, copper, which may be toxic to open ocean phytoplankton and bacteria, can form organic complexes. The formation of these complexes reduces the concentrations of bioavailable inorganic complexes of copper that could be toxic to sea life at high concentrations. Unlike copper, zinc toxicity in marine phytoplankton is low and there is no advantage to increasing the organic binding of Zn2+. In high nutrient-low chlorophyll regions, iron is the limiting nutrient, with the dominant species being strong organic complexes of Fe(III).[39]
See also
- Biogeochemical cycle
- Geochemical cycle
- Important publications in geochemistry
- Petrology
- Tephrochronology
References
- Albarède, Francis (2007). Geochemistry : an introduction. Translated from the French. (5th ed.). Cambridge: Cambridge Univ. Press. ISBN 9780521891486.
- McSween, Jr, Harry Y.; Huss, Gary R. (2010). Cosmochemistry. Cambridge University Press. ISBN 9781139489461.
- Kragh, Helge (2008). "From geochemistry to cosmochemistry: The origin of a scientific discipline, 1915–1955". In Reinhardt, Carsten (ed.). Chemical Sciences in the 20th Century: Bridging Boundaries. John Wiley & Sons. pp. 160–192. ISBN 978-3-527-30271-0.
- McSween, Jr., Harry Y.; Richardson, Steven M.; Uhle, Maria E. (2003). Geochemistry pathways and processes (2nd ed.). New York: Columbia University. ISBN 9780231509039.
- White, William M. Geochemistry (Unpublished). p. 1. Retrieved 14 March 2012.
- Mason, Brian (1992). Victor Moritz Goldschmidt : father of modern geochemistry. San Antonio, Tex.: Geochemical Society. ISBN 0-941809-03-X.
- "Welcome to GPS Geochemistry". GPS Research Program. California Institute of Technology. Retrieved 2 October 2017.
- Langmuir, Donald (1997). Aqueous environmental geochemistry. Upper Saddle River, N.J.: Prentice Hall. ISBN 9780023674129.
- Schlesinger, William H.; Bernhardt, Emily S. (2013). Biogeochemistry : an analysis of global change (Third ed.). Academic Press. ISBN 9780123858740.
- Kendall, Carol; Caldwell, Eric A. (1998). "Chapter 2: Fundamentals of Isotope Geochemistry". In Kendall, C.; McDonnell, J. J. (eds.). Isotope Tracers in Catchment Hydrology. Amsterdam: Elsevier Science. pp. 51–86. Retrieved 3 October 2017.
- Killops, Stephen D.; Killops, Vanessa J. (2013). Introduction to Organic Geochemistry. John Wiley & Sons. ISBN 9781118697207.
- Doane, TA (2017). "A survey of photogeochemistry". Geochem Trans. 18: 1. doi:10.1186/s12932-017-0039-y. PMC 5307419. PMID 28246525.
- Garrett, R.G.; Reimann, C.; Smith, D.B.; Xie, X. (November 2008). "From geochemical prospecting to international geochemical mapping: a historical overview: Table 1". Geochemistry: Exploration, Environment, Analysis. 8 (3–4): 205–217. doi:10.1144/1467-7873/08-174.
- McSween, Jr., Harry Y.; Huss, Gary R. (2010). Cosmochemistry. Cambridge University Press. ISBN 9781139489461.
- Olson, Gerald Schubert ; Donald L. Turcotte ; Peter (2001). Mantle convection in the earth and planets. Cambridge: Cambridge Univ. Press. ISBN 9780521798365.
- Wilson, Marjorie (2007). Igneous petrogenesis. Dordrecht: Springer. ISBN 9789401093880.
- Kendall, Carol; Caldwell, Eric A. (2000). "Chapter 2: Fundamentals of Isotope Geochemistry". In Kendall, Carol; McDonnell, J. J. (eds.). Isotope tracers in catchment hydrology. Amsterdam: Elsevier. pp. 51–86. ISBN 9780444501554. Retrieved 24 October 2017.
- Hoefs, Jochen (2015). "Isotope fractionation processes of selected elements". Stable Isotope Geochemistry: 47–134. doi:10.1007/978-3-319-19716-6_2. ISBN 978-3-319-19715-9.
- Lasaga, Antonio C.; Berner, Robert A. (April 1998). "Fundamental aspects of quantitative models for geochemical cycles". Chemical Geology. 145 (3–4): 161–175. Bibcode:1998ChGeo.145..161L. doi:10.1016/S0009-2541(97)00142-3.
- Data from table 6 of Cameron, A.G.W. (September 1973). "Abundances of the elements in the solar system". Space Science Reviews. 15 (1): 121. Bibcode:1973SSRv...15..121C. doi:10.1007/BF00172440.
- Palme, H.; Jones, A. (2003). "1.03 – Solar system abundance of the elements" (PDF). In Holland, H.D.; Turekian, K.K. (eds.). Treatise on Geochemistry. Volume 1: Meteorites, Comets and Planets (1st ed.). Oxford: Elsevier Science. pp. 41–61. doi:10.1016/B0-08-043751-6/01060-4. ISBN 9780080437514. Retrieved 3 October 2017.
- Lodders, Katharina (10 July 2003). "Solar System Abundances and Condensation Temperatures of the Elements". The Astrophysical Journal. 591 (2): 1220–1247. Bibcode:2003ApJ...591.1220L. CiteSeerX 10.1.1.695.5451. doi:10.1086/375492.
- Middlemost, Eric A. K. (2014). Magmas, Rocks and Planetary Development: A Survey of Magma/Igneous Rock Systems. Routledge. ISBN 9781317892649.
- Encrenaz, Therese; Bibring, Jean-Pierre; Blanc, M.; Barucci, Maria-Antonietta; Roques, Francoise; Zarka, Philippe (2004). The solar system (3rd ed.). Berlin: Springer. ISBN 9783540002413.
- Lewis, John (1995). Physics and Chemistry of the Solar System. Burlington: Elsevier Science. ISBN 9780323145848.
- Atreya, S.K; Mahaffy, P.R; Niemann, H.B; Wong, M.H; Owen, T.C (February 2003). "Composition and origin of the atmosphere of Jupiter—an update, and implications for the extrasolar giant planets". Planetary and Space Science. 51 (2): 105–112. Bibcode:2003P&SS...51..105A. doi:10.1016/S0032-0633(02)00144-7.
- Fortney, Jonathan (22 March 2010). "Viewpoint: Peering into Jupiter". Physics. 3: 26. doi:10.1103/Physics.3.26.
- Netburn, Deborah (15 September 2017). "As NASA's Cassini mission flames out over Saturn, scientists mark bittersweet end of mission". The Los Angeles Times. Retrieved 10 October 2017.
- Lang, Kenneth R. (2010). "11. Uranus and Neptune". NASA's Cosmos. Tufts University. Retrieved 11 October 2017.
- Anderson, Don L. (2007). New Theory of the Earth. Cambridge University Press. ISBN 9781139462082.
- "GRS". Jet Propulsion Laboratory. USA.gov. Retrieved 17 October 2017.
- Rhodes, Edgar A.; Evans, Larry G.; Nittler, Larry R.; Starr, Richard D.; Sprague, Ann L.; Lawrence, David J.; McCoy, Timothy J.; Stockstill-Cahill, Karen R.; Goldsten, John O.; Peplowski, Patrick N.; Hamara, David K.; Boynton, William V.; Solomon, Sean C. (December 2011). "Analysis of MESSENGER Gamma-Ray Spectrometer data from the Mercury flybys". Planetary and Space Science. 59 (15): 1829–1841. Bibcode:2011P&SS...59.1829R. doi:10.1016/j.pss.2011.07.018.
- Kieffer, Hugh H., ed. (1994). Mars (2nd ed.). Tucson: University of Arizona Press. ISBN 9780816512577.
- Morgan, John W.; Anders, Edward (December 1980). "Chemical composition of Earth, Venus, and Mercury". Proceedings of the National Academy of Sciences of the United States of America. 77 (12): 6973–6977. Bibcode:1980PNAS...77.6973M. doi:10.1073/pnas.77.12.6973. JSTOR 9538. PMC 350422. PMID 16592930.
-

- According to , which cites this: Klein, C., Hurlbut, C. S. (1993) Manual of Mineralogy, 21st Edition. John Wiley & Sons.
- Nameroff, T; Balistrieri, L; Murray, J (2002). "Suboxic Trace Metal Geochemistry in the Eastern Tropic North Pacific". Geochimica et Cosmochimica Acta. 66 (7): 1139–1158. Bibcode:2002GeCoA..66.1139N. doi:10.1016/s0016-7037(01)00843-2.
- Benjamin, M (2002). Water Chemistry. University of Washington. ISBN 1-57766-667-4.
- Bruland, K; Lohan, M (2003). "6.02 – Controls on Trace Metals in Seawater". In Holland, H.D.; Turekian, K.K. (eds.). Treatise on Geochemistry. Volume 6: The Oceans and Marine Geochemistry. pp. 23–47. Bibcode:2003TrGeo...6...23B. doi:10.1016/B0-08-043751-6/06105-3.
Further reading
- Faure, Gunter; Mensing, Teresa M. (2005). Isotopes : principles and applications (3rd ed.). New Jersey: Wiley. ISBN 0471384372.
- Holland, H.D.; Turekian, K.K., eds. (2003). Treatise on geochemistry (1st ed.). Oxford: Elsevier Science. ISBN 978-0-08-043751-4.
- Marshall, C.P.; Fairbridge, R.W., eds. (2006). Geochemistry. Berlin: SpringerLink. ISBN 1-4020-4496-8.
- Natural Environment Research Council. "Geochemistry data model". EarthDataModels.org. Retrieved 9 October 2017.
- Rollinson, Hugh R. (1996). Using geochemical data : evaluation, presentation, interpretation (Repr. ed.). Harlow: Longman. ISBN 978-0-582-06701-1.
- White, William M. Geochemistry (Unpublished). p. 1. Retrieved 14 March 2012.
External links
- The Geochemistry of Igneous Rocks (Gunn Interactive Ltd.)
| Overviews |  | |
|---|---|---|
| History of geology | ||
| Сomposition and structure | ||
| Historical geology | ||
| Motion | ||
| Water | ||
| Geophysics | ||
| Applications | ||
| Occupations | ||
| ||
Branches of chemistry | |
|---|---|
| Physical | |
| Organic | |
| Inorganic | |
| Analytical | |
| Others | |
| See also | |
| |