Hereditary elliptocytosis
Hereditary elliptocytosis, also known as ovalocytosis, is an inherited blood disorder in which an abnormally large number of the person's red blood cells are elliptical rather than the typical biconcave disc shape. Such morphologically distinctive erythrocytes are sometimes referred to as elliptocytes or ovalocytes. It is one of many red-cell membrane defects. In its severe forms, this disorder predisposes to haemolytic anaemia. Although pathological in humans, elliptocytosis is normal in camelids.
| Hereditary elliptocytosis | |
|---|---|
| Other names | Ovalocytosis, elliptocytes, ovalocytes |
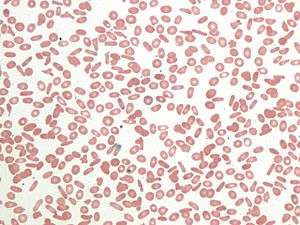 | |
| Peripheral blood smear showing an abundant number of elliptocytes | |
| Specialty | Hematology |
Pathophysiology
Common hereditary elliptocytosis
A number of genes have been linked to common hereditary elliptocytosis (many involve the same gene as forms of Hereditary spherocytosis, or HS):
| Type | OMIM | Gene |
|---|---|---|
| EL1 or HS5 | 611804 | EPB41 |
| EL2 or HS3 | 130600 | SPTA1 |
| EL3 or HS2 | 182870 | SPTB |
| EL4 or HS4 or SEO | 109270 | SLC4A1 |
These mutations have a common end result; they destabilise the cytoskeletal scaffold of cells. This stability is especially important in erythrocytes as they are constantly under the influence of deforming shear forces. As disc-shaped erythrocytes pass through capillaries, which can be 2-3 micrometres wide, they are forced to assume an elliptical shape in order to fit through. Normally, this deformation lasts only as long as a cell is present in a capillary, but in hereditary elliptocytosis the instability of the cytoskeleton means that erythrocytes deformed by passing through a capillary are forever rendered elliptical. These elliptical cells are taken up by the spleen and removed from circulation when they are younger than they would normally be, meaning that the erythrocytes of people with hereditary elliptocytosis have a shorter than average life-span (a normal person's erythrocytes average 120 days or more).
.JPG)
- EL2 and EL3: The most common genetic defects (present in two-thirds of all cases of hereditary elliptocytosis) are in genes for the polypeptides α-spectrin or β-spectrin. These two polypeptides combine with one another in vivo to form an αβ heterodimer. These αβ heterodimers then combine to form spectrin tetramers. These spectrin tetramers are among the basic structural subunits of the cytoskeleton of all cells in the body. Although there is much interindividual variability, it is generally true that α-spectrin mutations result in an inability of α-spectrin to interact properly with β-spectrin to form a heterodimer. In contrast, it is generally true that β-spectrin mutations lead to αβ heterodimers being incapable of combining to form spectrin tetramers.[1] In both cases the end result is a weakness in the cytoskeleton of the cell. Individuals with a single mutation in one of the spectrin genes are usually asymptomatic, but those who are homozygotes or are compound heterozygotes (i.e. they are heterozygous for two different elliptocytosis-causing mutations) have sufficient cell membrane instability to have a clinically significant haemolytic anaemia.
- EL1: Less common than spectrin mutations are band 4.1 mutations. Spectrin tetramers must bind to actin in order to create a proper cytoskeleton scaffold, and band 4.1 is an important protein involved in the stabilisation of the link between spectrin and actin. Similarly to the spectrin mutations, band 4.1 mutations cause a mild haemolytic anaemia in the heterozygous state, and a severe haemolytic disease in the homozygous state.
- EL4: Southeast Asian ovalocytosis is associated with the Band 3 protein.
- Another group of mutations that lead to elliptocytosis are those that cause glycophorin C deficiencies. There are three phenotypes caused by abnormal glycophorin C, these are named Gerbich, Yus and Leach (see glycophorin C for more information). Only the rarest of the three, the Leach phenotype, causes elliptocytosis. Glycophorin C has the function of holding band 4.1 to the cell membrane. It is thought that elliptocytosis in glycophorin C deficiency is actually the consequence of a band 4.1 deficit, as glycophorin C deficient individuals also have reduced intracellular band 4.1 (probably due to the reduced number of binding sites for band 4.1 in the absence of glycoprotein C).
Inheritance of multiple mutations tends to infer more serious disease. For instance, the most common genotype responsible for HPP occurs when the affected individual inherits an α-spectrin mutation from one parent (i.e. one parent has hereditary elliptocytosis) and the other parent passes on an as-yet-undefined defect that causes the affected individual's cells to preferentially produce the defective α-spectrin rather than normal α-spectrin.
Diagnosis
The diagnosis of hereditary elliptocytosis is usually made by coupling a family history of the condition with an appropriate clinical presentation and confirmation on a blood smear. In general it requires that at least 25% of erythrocytes in the specimen are abnormally elliptical in shape, though the observed percentage of elliptocytes can be 100%. This is in contrast to the rest of the population, in which it is common for up to 15% of erythrocytes to be elliptical.[2]
If some doubt remains regarding the diagnosis, definitive diagnosis can involve osmotic fragility testing, an autohaemolysis test, and direct protein assaying by gel electrophoresis.[3]
Treatment
The vast majority of those with hereditary elliptocytosis require no treatment whatsoever. They have a mildly increased risk of developing gallstones, which is treated surgically with a cholecystectomy if pain becomes problematic. This risk is relative to the severity of the disease.
Folate helps to reduce the extent of haemolysis in those with significant haemolysis due to hereditary elliptocytosis.
Because the spleen breaks down old and worn-out blood cells, those individuals with more severe forms of hereditary elliptocytosis can have splenomegaly. Symptoms of splenomegaly can include:
- Vague, poorly localised abdominal pain
- Fatigue and dyspnoea
- Growth failure
- Leg ulcers
- Gallstones.
Removal of the spleen (splenectomy) is effective in reducing the severity of these complications, but is associated with an increased risk of overwhelming bacterial septicaemia, and is only performed on those with significant complications. Because many neonates with severe elliptocytosis progress to have only a mild disease, and because this age group is particularly susceptible to pneumococcal infections, a splenectomy is only performed on those under 5 years old when it is absolutely necessary.
Prognosis
Those with hereditary elliptocytosis have a good prognosis, only those with very severe disease have a shortened life expectancy.
Epidemiology
The incidence of hereditary elliptocytosis is hard to determine, as many sufferers of the milder forms of the disorder are asymptomatic and their condition never comes to medical attention.[4] Around 90% of those with this disorder are thought to fall into the asymptomatic population. It is estimated that its incidence is between 3 and 5 per 10,000 in the United States,[5] and that those of African and Mediterranean descent are of higher risk. Because it can confer resistance to malaria, some subtypes of hereditary elliptocytosis are significantly more prevalent in regions where malaria is endemic. For example, in equatorial Africa its incidence is estimated at 60-160 per 10,000,[6] and in Malayan natives its incidence is 1500-2000 per 10,000.[7] Almost all forms of hereditary elliptocytosis are autosomal dominant, and both sexes are therefore at equal risk of having the condition. The most important exception to this rule of autosomal dominance is for a subtype of hereditary elliptocytosis called hereditary pyropoikilocytosis (HPP), which is autosomal recessive.[8]
There are three major forms of hereditary elliptocytosis: common hereditary elliptocytosis, spherocytic elliptocytosis and southeast Asian ovalocytosis.
Common hereditary elliptocytosis is the most common form of elliptocytosis, and the form most extensively researched. Even when looking only at this form of elliptocytosis, there is a high degree of variability in the clinical severity of its subtypes. A clinically significant haemolytic anaemia occurs only in 5-10% of sufferers, with a strong bias towards those with more severe subtypes of the disorder.
Southeast Asian ovalocytosis and spherocytic elliptocytosis are less common subtypes predominantly affecting those of south-east Asian and European ethnic groups, respectively.
The following categorisation of the disorder demonstrates its heterogeneity:[9]
- Common hereditary elliptocytosis (in approximate order from least severe to most severe)
- With asymptomatic carrier status - individuals have no symptoms of disease and diagnosis is only able to be made on blood film
- With mild disease - individuals have no symptoms, with a mild and compensated haemolytic anaemia
- With sporadic haemolysis - individuals are at risk of haemolysis in the presence of particular comorbidities, including infections, and vitamin B12 deficiency
- With neonatal poikilocytosis - individuals have a symptomatic haemolytic anaemia with poikilocytosis that resolves in the first year of life
- With chronic haemolysis - individual has a moderate to severe symptomatic haemolytic anaemia (this subtype has variable penetrance in some pedigrees)
- With homozygosity or compound heterozygosity - depending on the exact mutations involved, individuals may lie anywhere in the spectrum between having a mild haemolytic anaemia and having a life-threatening haemolytic anaemia with symptoms mimicking those of HPP (see below)
- With pyropoikilocytosis (HPP) - individuals are typically of African descent and have a life-threateningly severe haemolytic anaemia with micropoikilocytosis (small and misshapen erythrocytes) that is compounded by a marked instability of erythrocytes in even mildly elevated temperatures (pyropoikilocytosis is often found in burns victims and is the term is commonly used in reference to such people)
- South-east Asian ovalocytosis (SAO) (also called stomatocytic elliptocytosis) - individuals are of South-East Asian descent (typically Malaysian, Indonesian, Melanesian, New Guinean or Filipino, have a mild haemolytic anaemia, and has increased resistance to malaria
- Spherocytic elliptocytosis (also called hereditary haemolytic ovalocytosis) - individuals are of European descent and elliptocytes and spherocytes are simultaneously present in their blood
History
Elliptocytosis was first described in 1904,[10] and was first recognised as a hereditary condition in 1932.[11] More recently it has become clear that the severity of the condition is highly variable,[12] and there is much genetic variability amongst those affected.[13]
See also
References
- McMullin MF (1999). "The molecular basis of disorders of the red cell membrane". J. Clin. Pathol. 52 (4): 245–8. doi:10.1136/jcp.52.4.245. PMC 501324. PMID 10474512.
- Gerard M. Doherty (2010). Current Diagnosis & Treatment - Surgery (13th ed.). McGraw Hill Professional. pp. 204–5. ISBN 978-0-07-163515-8. Retrieved 5 May 2011.
- Robert S. Hillman; Kenneth A. Ault; Henry M. Rinder (2005). Hematology in clinical practice: a guide to diagnosis and management (4th ed.). McGraw-Hill Professional. p. 147. ISBN 978-0-07-144035-6. Retrieved 5 May 2011.
- Kim, D (24 May 2006). "Elliptocytosis, Hereditary". Medscape. WebMD LLC. Retrieved 12 August 2013.
- Bannerman, Rm; Renwick, Jh (July 1962). "The hereditary elliptocytoses: clinical and linkage data". Annals of Human Genetics. 26 (1): 23–38. doi:10.1111/j.1469-1809.1962.tb01306.x. ISSN 0003-4800. PMID 13864689.
- Hoffman, R; Benz, E; Shattil, S; Furie, B; Cohen, H (2005). Hoffman Hematology: Basic Principles and Practice (4th ed.). Philadelphia: Churchill Livingstone. ISBN 978-0-443-06628-3.
- Cattani, Ja; Gibson, Fd; Alpers, Mp; Crane, Gg (1987). "Hereditary ovalocytosis and reduced susceptibility to malaria in Papua New Guinea" (Free full text). Transactions of the Royal Society of Tropical Medicine and Hygiene. 81 (5): 705–9. doi:10.1016/0035-9203(87)90001-0. ISSN 0035-9203. PMID 3329776.
- Kutlar, A (22 October 2013). "Hereditary Pyropoikilocytosis". Medscape. WebMD LLC. Retrieved 29 January 2015.
- Coetzer T, Lawler J, Prchal JT, Palek J (1 September 1987). "Molecular determinants of clinical expression of hereditary elliptocytosis and pyropoikilocytosis". Blood. 70 (3): 766–72. doi:10.1182/blood.V70.3.766.766. PMID 3620700.
- Dresbach M (1904). "Elliptical human red corpuscles". Science. 19 (481): 469–470. Bibcode:1904Sci....19..469D. doi:10.1126/science.19.481.469. PMID 17730874.
- Hunter, WC (1932). "Further study of a white family showing elliptical erythrocytes". Ann Intern Med. 6 (6): 775–781. doi:10.7326/0003-4819-6-6-775.
- Gallagher, Pg (2005). "Red Cell Membrane Disorders". Hematology. 2005 (1): 13–8. doi:10.1182/asheducation-2005.1.13. PMID 16304353.
- Tse, Wt; Lux, Se (January 1999). "Red blood cell membrane disorders". British Journal of Haematology. 104 (1): 2–13. doi:10.1111/j.1365-2141.1999.01130.x. ISSN 0007-1048. PMID 10027705.
External links
| Classification | |
|---|---|
| External resources |
- Hereditary Elliptocytosis Image of hereditary elliptocytosis
- MedlinePlus Entry
{kind=link}