Alpha-galactosidase
Alpha-galactosidase (α-GAL, also known as α-GAL A; E.C. 3.2.1.22) is a glycoside hydrolase enzyme that hydrolyses the terminal alpha-galactosyl moieties from glycolipids and glycoproteins. Glycosidase is an important class of enzyme catalyzing many catabolic processes, including cleaving glycoproteins and glycolipids, and polysaccharides. Specifically, α-GAL catalyzes the removal of the terminal α-galactose from oligosaccharides.[5]



| alpha-galactosidase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 Alpha-galactosidase tetramer, Mortierella vinacea | |||||||||
| Identifiers | |||||||||
| EC number | 3.2.1.22 | ||||||||
| CAS number | 9025-35-8 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
The enzyme is encoded by the GLA gene.[6] Two recombinant forms of human alpha-galactosidase are called agalsidase alpha (INN) and agalsidase beta (INN). A mold-derived form is the primary ingredient in gas relief supplements.
Function
This enzyme is a homodimeric glycoprotein that hydrolyses the terminal alpha-galactosyl moieties from glycolipids and glycoproteins. It predominantly hydrolyzes ceramide trihexoside, and it can catalyze the hydrolysis of melibiose into galactose and glucose.
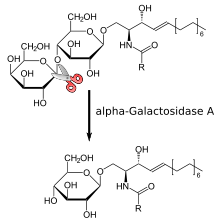
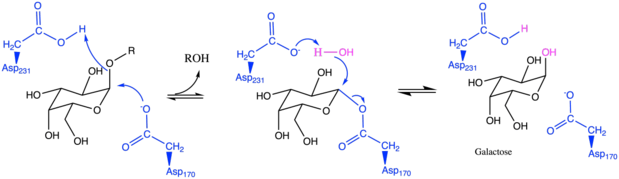
Reaction mechanism
Disease relevance
Fabry disease
Signs and Symptoms
Defects in human α-GAL result in Fabry disease, a rare lysosomal storage disorder and sphingolipidosis that results from a failure to catabolize α-D-galactosyl glycolipid moieties.[10] Characteristic features include episodes of pain in hands and feet (acroparethesias), dark red spots on skin (angiokeratomas), decreased sweating (hypohidrosis), decreased vision (corneal opacity), gastrointestinal problems, hearing loss, tinnitus, etc.. Complications for this disease can be life-threatening and may include progressive kidney damage, heart attack, and stroke. This disease may have late onset and only affect the heart or kidneys.[11]
Fabry disease is an X-linked disease, affecting 1 in 40,000 males. However, unlike other X-linked diseases, this condition also creates significant medical problems for females carrying only 1 copy of the defective GLA gene. These women may experience many classic symptoms of the disorder including cardiac and kidney problems. However, a small number of females carrying only one copy of the mutated GLA gene never shows any symptoms of Fabry disease.
Cause
Mutations to the GLA gene encoding α-GAL may result in complete loss of function of the enzyme. α-GAL is a lysosomal protein responsible for breaking down globotriaosylceramide, a fatty substance stored various types of cardiac and renal cells.[12] When globotriaosylceramide is not properly catabolized, it is accumulated in cells lining blood vessels in the skin, cells in the kidney, heart and nervous system. As a result, signs and symptoms of Fabry disease begin to manifest.[11]
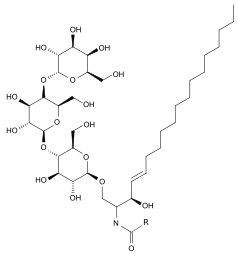
Treatment
There are two treatment options for Fabry disease: recombinant enzyme replacement therapy and pharmacological chaperone therapy.
Recombinant enzyme replacement therapy (RERT)
RERT was approved as a treatment for Fabry disease in the United States in 2003.[13][14][15]
Two recombinant enzyme replacement therapies are available to functionally compensate for alpha-galactosidase deficiency. Agalsidase alpha and beta are both recombinant forms of the human α-galactosidase A enzyme and both have the same amino acid sequence as the native enzyme. Agalsidase alpha and beta differ in the structures of their oligosaccharide side chains.[16]
In Fabry disease patients, 88% percent of patients develop IgG antibodies towards the injected recombinant enzyme, as it is foreign to their immune system. One suggested approach to solving this problem involves converting the paralogous enzyme α-NAGAL (NAGA) into one that has with α-GAL activity. Because patients still have a function NAGA gene, their immune system will not produce NAGA antibodies.[17]
Agalsidase alpha
The pharmaceutical company Shire manufactures agalsidase alfa (INN) under the trade name Replagal as a treatment for Fabry disease,[18] and was granted marketing approval in the EU in 2001.[19] FDA approval was applied for the United States.[20] However, in 2012, Shire withdrew their application for approval in the United States citing that the agency will require additional clinical trials before approval.[21]
Agalsidase beta
The pharmaceutical company Genzyme produces synthetic agalsidase beta (INN) under the trade name Fabrazyme for treatment of Fabry disease. In 2009, contamination at Genzyme's Allston, Massachusetts plant caused a worldwide shortage of Fabrazyme, and supplies were rationed to patients at one-third the recommended dose. Some patients have petitioned to break the company's patent on the drug under the "march-in" provisions of the Bayh–Dole Act.[20]
Pharmacological chaperone therapy
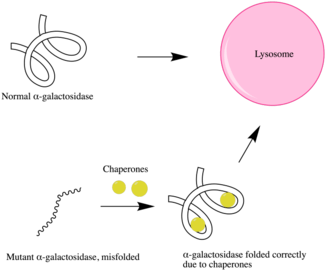
Fabry patients who display neurological symptoms cannot receive RERT because recombinant enzymes cannot normally pass the blood-brain barrier. Thus, a more suitable alternative treatment is used: pharmacological chaperone therapy.
It has been shown that more potent competitive inhibitors of an enzyme can act as a more powerful chemical chaperone for the corresponding mutant enzyme that fails to maintain proper folding and conformation, despite its intact active site. These chemical chaperones bind to the active site of the mutant enzyme, which can help promote proper folding and stabilize the mutant enzyme. Thus, this results in functional mutant enzymes that will not be degraded via the ubiquitin-proteasome pathway.
1-Deoxygalactonojirimycin (DGJ) has been shown to be both a potent competitive inhibitor of α-GAL and an effective chaperone to for Fabry disease, increasing intracellular α-GAL's activity by 14-fold.[24][25]
Modifying blood type group B to group O
α-GAL, known as B-zyme in this context, has also demonstrated its ability to convert human blood group B to human blood group O, which can be transfused to patients of all blood types in the ABO blood group categorization. The current B-zyme used comes from Bacteroides fragilis.[23] The idea of maintaining a blood supply at healthcare facilities with all non-O units converted to O units is achieved using enzyme-converted to group O technology, first developed in 1982.[26]
Advantages
A blood bank with ECO blood demonstrates the following advantages:[27]
- Compatible with and transfusable to patients of all blood groups
- Reduce the demand for specific ABO blood groups A, B, AB
- Reduce cost of maintaining a blood bank inventory in hospitals
- Reduce blood transfusion reactions due to human error and ABO incompatibility
- Reduce wastage of less needed blood types
Mechanism of Action
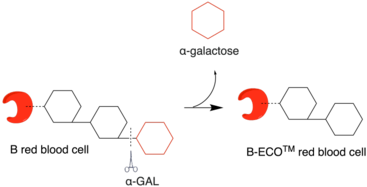
Red blood cell (RBC) surfaces are decorated with the glycoproteins and glycolipids that have the same basic sequence with terminal sugar α1‐2‐linked fucose linked to the penultimate galactose. This galactose molecule is called the H antigen.[28][29][30] Blood type A, B, AB, and O differ only in the sugar (red molecule in the illustration) linked with the penultimate galactose. For blood type B, this linked sugar is an α-1‐3‐linked galactose. Using α-GAL, this terminal galactose molecule can be removed, converting RBC to type O.
Supplements
α-GAL derived from aspergillus niger (a common mold) is an active ingredient in products marketed to reduce stomach gas production after eating foods known to cause gas. It is optimally active at 55 degrees C, after which its half-life is 120 minutes.[31]
There are scores of supplements containing the enzyme over the counter in the United States and many more world wide. Products with Alpha-galactosidase include:
- Beano
- CVS BeanAid
- Enzymedica's BeanAssist
- Gasfix
- Bloateez (in India as Cogentrix)
See also
- Beta-galactosidase
- Migalastat, a drug targeting alpha-galactosidase
- Classification of α-galactosidases (according to CAZy)
References
- GRCh38: Ensembl release 89: ENSG00000102393 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000031266 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Scriver CR, Sly WS, Childs B, ABeaudet AL, Valle D, Kinzler KW, Vogelstein B (15 December 2000). The Metabolic & Molecular Basis of Inherited Disease (8th ed.). McGraw-Hill. ISBN 978-0-07-913035-8.
- Calhoun DH, Bishop DF, Bernstein HS, Quinn M, Hantzopoulos P, Desnick RJ (November 1985). "Fabry disease: isolation of a cDNA clone encoding human alpha-galactosidase A". Proceedings of the National Academy of Sciences of the United States of America. 82 (21): 7364–8. Bibcode:1985PNAS...82.7364C. doi:10.1073/pnas.82.21.7364. PMC 391345. PMID 2997789.
- Koshland DE (1953). "Stereochemistry and the Mechanism of Enzymatic Reactions". Biological Reviews. 28 (4): 416–436. doi:10.1111/j.1469-185x.1953.tb01386.x.
- Brumer H, Sims PF, Sinnott ML (April 1999). "Lignocellulose degradation by Phanerochaete chrysosporium: purification and characterization of the main alpha-galactosidase". The Biochemical Journal. 339 ( Pt 1) (1): 43–53. doi:10.1042/bj3390043. PMC 1220126. PMID 10085226.
- Vocadlo DJ, Davies GJ (October 2008). "Mechanistic insights into glycosidase chemistry". Current Opinion in Chemical Biology. 12 (5): 539–55. doi:10.1016/j.cbpa.2008.05.010. PMID 18558099.
- "Entrez Gene: GLA galactosidase, alpha".
- Reference. "Fabry disease". Genetics Home Reference. Retrieved 2019-03-09.
- Ronco C, Bellomo R, Bellasi A (2019). Critical Care Nephrology (Third ed.). Elsevier. pp. 704–711.e2. doi:10.1016/B978-0-323-44942-7.00115-1 (inactive 2020-05-21). ISBN 978-0-323-44942-7.
- Schiffmann R, Kopp JB, Austin HA, Sabnis S, Moore DF, Weibel T, Balow JE, Brady RO (June 2001). "Enzyme replacement therapy in Fabry disease: a randomized controlled trial". JAMA. 285 (21): 2743–9. doi:10.1001/jama.285.21.2743. PMID 11386930.
- Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ (July 2001). "Safety and efficacy of recombinant human alpha-galactosidase A replacement therapy in Fabry's disease". The New England Journal of Medicine. 345 (1): 9–16. doi:10.1056/nejm200107053450102. PMID 11439963.
- Desnick RJ, Schuchman EH (December 2002). "Enzyme replacement and enhancement therapies: lessons from lysosomal disorders". Nature Reviews. Genetics. 3 (12): 954–66. doi:10.1038/nrg963. PMID 12459725.
- Fervenza FC, Torra R, Warnock DG (December 2008). "Safety and efficacy of enzyme replacement therapy in the nephropathy of Fabry disease". Biologics. 2 (4): 823–43. doi:10.2147/btt.s3770. PMC 2727881. PMID 19707461.
- Tomasic IB, Metcalf MC, Guce AI, Clark NE, Garman SC (July 2010). "Interconversion of the specificities of human lysosomal enzymes associated with Fabry and Schindler diseases". The Journal of Biological Chemistry. 285 (28): 21560–6. doi:10.1074/jbc.M110.118588. PMC 2898384. PMID 20444686.
- Keating GM (October 2012). "Agalsidase alfa: a review of its use in the management of Fabry disease". BioDrugs. 26 (5): 335–54. doi:10.2165/11209690-000000000-00000. PMID 22946754.
- "Shire Submits Biologics License Application (BLA) for REPLAGAL with the U.S. Food and Drug Administration (FDA)". FierceBiotech.
- "With A Life-Saving Medicine In Short Supply, Patients Want Patent Broken". 2010-08-04. Archived from the original on 14 September 2010. Retrieved 2010-09-02.
- Grogan K (2012-03-15). "Shire withdraws Replagal in USA as FDA wants more trials". PharmaTimes. Archived from the original on 2014-08-19.
- Frustaci A, Chimenti C, Ricci R, Natale L, Russo MA, Pieroni M, Eng CM, Desnick RJ (July 2001). "Improvement in cardiac function in the cardiac variant of Fabry's disease with galactose-infusion therapy". The New England Journal of Medicine. 345 (1): 25–32. doi:10.1056/nejm200107053450104. PMID 11439944.
- Liu QP, Sulzenbacher G, Yuan H, Bennett EP, Pietz G, Saunders K, et al. (April 2007). "Bacterial glycosidases for the production of universal red blood cells". Nature Biotechnology. 25 (4): 454–64. doi:10.1038/nbt1298. PMID 17401360.
- Asano N, Ishii S, Kizu H, Ikeda K, Yasuda K, Kato A, Martin OR, Fan JQ (July 2000). "In vitro inhibition and intracellular enhancement of lysosomal alpha-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives". European Journal of Biochemistry. 267 (13): 4179–86. doi:10.1046/j.1432-1327.2000.01457.x. PMID 10866822.
- Fan JQ, Ishii S, Asano N, Suzuki Y (January 1999). "Accelerated transport and maturation of lysosomal alpha-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor". Nature Medicine. 5 (1): 112–5. doi:10.1038/4801. PMID 9883849.
- Goldstein J, Siviglia G, Hurst R, Lenny L, Reich L (January 1982). "Group B erythrocytes enzymatically converted to group O survive normally in A, B, and O individuals". Science. 215 (4529): 168–70. Bibcode:1982Sci...215..168G. doi:10.1126/science.6274021. PMID 6274021.
- Olsson ML, Clausen H (January 2008). "Modifying the red cell surface: towards an ABO-universal blood supply". British Journal of Haematology. 140 (1): 3–12. doi:10.1111/j.1365-2141.2007.06839.x. PMID 17970801.
- Watkins WM (1980). "Biochemistry and Genetics of the ABO, Lewis, and P blood group systems". Advances in Human Genetics. Springer US. 10: 1–136, 379–85. doi:10.1007/978-1-4615-8288-5_1. ISBN 9781461582908. PMID 6156588.
- Oriol R, Le Pendu J, Mollicone R (1986). "Genetics of ABO, H, Lewis, X and related antigens". Vox Sanguinis. 51 (3): 161–71. doi:10.1111/j.1423-0410.1986.tb01946.x. PMID 2433836.
- Clausen H, Hakomori S (1989). "ABH and related histo-blood group antigens; immunochemical differences in carrier isotypes and their distribution". Vox Sanguinis. 56 (1): 1–20. doi:10.1159/000460912. PMID 2464874.
- Patil AG, K PK, Mulimani VH, Veeranagouda Y, Lee K (November 2010). "alpha-Galactosidase from Bacillus megaterium VHM1 and its application in removal of flatulence-causing factors from soymilk". Journal of Microbiology and Biotechnology. 20 (11): 1546–54. doi:10.4014/jmb.0912.12012. PMID 21124061.
Further reading
- Naumov DG (2004). "[Phylogenetic analysis of alpha-galactosidases of the GH27 family]". Molekuliarnaia Biologiia (in Russian). 38 (3): 463–76. PMID 15285616. Republished as: Naumoff DG (2004). "Phylogenetic Analysis of α-Galactosidases of the GH27 Family". Molecular Biology. 38 (3): 388–400. doi:10.1023/B:MBIL.0000032210.97006.de.
- Eng CM, Desnick RJ (1994). "Molecular basis of Fabry disease: mutations and polymorphisms in the human alpha-galactosidase A gene". Human Mutation. 3 (2): 103–11. doi:10.1002/humu.1380030204. PMID 7911050.
- Caillaud C, Poenaru L (2002). "[Gaucher's and Fabry's diseases: biochemical and genetic aspects]" [Gaucher's and Fabry's diseases: biochemical and genetic aspects]. Journal de la Société de Biologie (in French). 196 (2): 135–40. doi:10.1051/jbio/2002196020135. PMID 12360742. INIST:13891620.
- Germain DP (2002). "[Fabry's disease (alpha-galactosidase-A deficiency): physiopathology, clinical signs, and genetic aspects]" [Fabry's disease (alpha-galactosidase-A deficiency): physiopathology, clinical signs, and genetic aspects]. Journal de la Société de Biologie (in French). 196 (2): 161–73. doi:10.1051/jbio/2002196020161. PMID 12360745. INIST:13891623.
- Schaefer E, Mehta A, Gal A (March 2005). "Genotype and phenotype in Fabry disease: analysis of the Fabry Outcome Survey". Acta Paediatrica. 94 (447): 87–92, discussion 79. doi:10.1080/08035320510031045. PMID 15895718.
- Levin M (January 2006). "Fabry disease". Drugs of Today. 42 (1): 65–70. doi:10.1358/dot.2006.42.1.957357. PMID 16511611.
- Lidove O, Joly D, Barbey F, Bekri S, Alexandra JF, Peigne V, Jaussaud R, Papo T (February 2007). "Clinical results of enzyme replacement therapy in Fabry disease: a comprehensive review of literature". International Journal of Clinical Practice. 61 (2): 293–302. doi:10.1111/j.1742-1241.2006.01237.x. PMID 17263716.
- Dean KJ, Sweeley CC (October 1979). "Studies on human liver alpha-galactosidases. I. Purification of alpha-galactosidase A and its enzymatic properties with glycolipid and oligosaccharide substrates". The Journal of Biological Chemistry. 254 (20): 9994–10000. PMID 39940.
- Ishii S, Sakuraba H, Suzuki Y (April 1992). "Point mutations in the upstream region of the alpha-galactosidase A gene exon 6 in an atypical variant of Fabry disease". Human Genetics. 89 (1): 29–32. doi:10.1007/BF00207037. PMID 1315715.
- Ioannou YA, Bishop DF, Desnick RJ (December 1992). "Overexpression of human alpha-galactosidase A results in its intracellular aggregation, crystallization in lysosomes, and selective secretion". The Journal of Cell Biology. 119 (5): 1137–50. doi:10.1083/jcb.119.5.1137. PMC 2289730. PMID 1332979.
- von Scheidt W, Eng CM, Fitzmaurice TF, Erdmann E, Hübner G, Olsen EG, Christomanou H, Kandolf R, Bishop DF, Desnick RJ (February 1991). "An atypical variant of Fabry's disease with manifestations confined to the myocardium". The New England Journal of Medicine. 324 (6): 395–9. doi:10.1056/NEJM199102073240607. PMID 1846223.
- Koide T, Ishiura M, Iwai K, Inoue M, Kaneda Y, Okada Y, Uchida T (January 1990). "A case of Fabry's disease in a patient with no alpha-galactosidase A activity caused by a single amino acid substitution of Pro-40 by Ser". FEBS Letters. 259 (2): 353–6. doi:10.1016/0014-5793(90)80046-L. PMID 2152885.
- Kornreich R, Bishop DF, Desnick RJ (June 1990). "Alpha-galactosidase A gene rearrangements causing Fabry disease. Identification of short direct repeats at breakpoints in an Alu-rich gene". The Journal of Biological Chemistry. 265 (16): 9319–26. PMID 2160973.
- Sakuraba H, Oshima A, Fukuhara Y, Shimmoto M, Nagao Y, Bishop DF, Desnick RJ, Suzuki Y (November 1990). "Identification of point mutations in the alpha-galactosidase A gene in classical and atypical hemizygotes with Fabry disease". American Journal of Human Genetics. 47 (5): 784–9. PMC 1683686. PMID 2171331.
- Bernstein HS, Bishop DF, Astrin KH, Kornreich R, Eng CM, Sakuraba H, Desnick RJ (April 1989). "Fabry disease: six gene rearrangements and an exonic point mutation in the alpha-galactosidase gene". The Journal of Clinical Investigation. 83 (4): 1390–9. doi:10.1172/JCI114027. PMC 303833. PMID 2539398.
- Kornreich R, Desnick RJ, Bishop DF (April 1989). "Nucleotide sequence of the human alpha-galactosidase A gene". Nucleic Acids Research. 17 (8): 3301–2. doi:10.1093/nar/17.8.3301. PMC 317741. PMID 2542896.
- Bishop DF, Kornreich R, Desnick RJ (June 1988). "Structural organization of the human alpha-galactosidase A gene: further evidence for the absence of a 3' untranslated region". Proceedings of the National Academy of Sciences of the United States of America. 85 (11): 3903–7. Bibcode:1988PNAS...85.3903B. doi:10.1073/pnas.85.11.3903. PMC 280328. PMID 2836863.
- Quinn M, Hantzopoulos P, Fidanza V, Calhoun DH (1987). "A genomic clone containing the promoter for the gene encoding the human lysosomal enzyme, alpha-galactosidase A". Gene. 58 (2–3): 177–88. doi:10.1016/0378-1119(87)90374-X. PMID 2892762.
- Bishop DF, Calhoun DH, Bernstein HS, Hantzopoulos P, Quinn M, Desnick RJ (July 1986). "Human alpha-galactosidase A: nucleotide sequence of a cDNA clone encoding the mature enzyme". Proceedings of the National Academy of Sciences of the United States of America. 83 (13): 4859–63. Bibcode:1986PNAS...83.4859B. doi:10.1073/pnas.83.13.4859. PMC 323842. PMID 3014515.
- Lemansky P, Bishop DF, Desnick RJ, Hasilik A, von Figura K (February 1987). "Synthesis and processing of alpha-galactosidase A in human fibroblasts. Evidence for different mutations in Fabry disease". The Journal of Biological Chemistry. 262 (5): 2062–5. PMID 3029062.
- Tsuji S, Martin BM, Kaslow DC, Migeon BR, Choudary PV, Stubbleflied BK, Mayor JA, Murray GJ, Barranger JA, Ginns EI (June 1987). "Signal sequence and DNA-mediated expression of human lysosomal alpha-galactosidase A". European Journal of Biochemistry. 165 (2): 275–80. doi:10.1111/j.1432-1033.1987.tb11438.x. PMID 3036505.
External links
- alpha-Galactosidase at the US National Library of Medicine Medical Subject Headings (MeSH)
- Human GLA genome location and GLA gene details page in the UCSC Genome Browser.
This article incorporates text from the United States National Library of Medicine, which is in the public domain.
PDB gallery | |
|---|---|
|


{kind=link}