Glycogen debranching enzyme
A debranching enzyme is a molecule that helps facilitate the breakdown of glycogen, which serves as a store of glucose in the body, through glucosyltransferase and glucosidase activity. Together with phosphorylases, debranching enzymes mobilize glucose reserves from glycogen deposits in the muscles and liver. This constitutes a major source of energy reserves in most organisms. Glycogen breakdown is highly regulated in the body, especially in the liver, by various hormones including insulin and glucagon, to maintain a homeostatic balance of blood-glucose levels.[5] When glycogen breakdown is compromised by mutations in the glycogen debranching enzyme, metabolic diseases such as Glycogen storage disease type III can result.[6][7]


| 4-α-glucanotransferase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC number | 2.4.1.25 | ||||||||
| CAS number | 9032-09-1 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
| amylo-α-1,6-glucosidase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| EC number | 3.2.1.33 | ||||||||
| CAS number | 9012-47-9 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
Glucosyltransferase and glucosidase are performed by a single enzyme in mammals, yeast, and some bacteria, but by two distinct enzymes in E. coli and other bacteria, complicating nomenclature. Proteins that catalyze both functions are referred to as glycogen debranching enzymes (GDEs). When glucosyltransferase and glucosidase are catalyzed by distinct enzymes, "glycogen debranching enzyme" usually refers to the glucosidase enzyme. In some literature, an enzyme capable only of glucosidase is referred to as a "debranching enzyme".[8]
Function
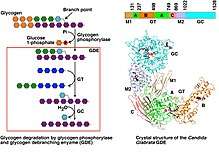
Together with phosphorylase, glycogen debranching enzymes function in glycogen breakdown and glucose mobilization. When phosphorylase has digested a glycogen branch down to four glucose residues, it will not remove further residues. Glycogen debranching enzymes assist phosphorylase, the primary enzyme involved in glycogen breakdown, mobilize glycogen stores. Phosphorylase can only cleave α-1,4- glycosidic bond between adjacent glucose molecules in glycogen but branches also exist as α-1,6 linkages. When phosphorylase reaches four residues from a branching point it stops cleaving; because 1 in 10 residues is branched, cleavage by phosphorylase alone would not be sufficient in mobilizing glycogen stores.[9][10] Before phosphorylase can resume catabolism, debranching enzymes perform two functions:
- 4-α-D-glucanotransferase (EC 2.4.1.25), or glucosyltransferase, transfers three glucose residues from the four-residue glycogen branch to a nearby branch. This exposes a single glucose residue joined to the glucose chain through an α -1,6 glycosidic linkage[9]
- Amylo-α-1,6-glucosidase (EC 3.2.1.33), or glucosidase, cleaves the remaining alpha-1,6 linkage, producing glucose and a linear chain of glycogen.[9] The mechanism by which the glucosidase cleaves the α -1,6-linkage is not fully known because the amino acids in the active site have not yet been identified. It is thought to proceed through a two step acid base assistance type mechanism, with an oxocarbenium ion intermediate, and retention of configuration in glucose.[11] This is a common method through which to cleave bonds, with an acid below the site of hydrolysis to lend a proton and a base above to deprotinate a water which can then act as a nucleophile. These acids and bases are amino acid side chains in the active site of the enzyme. A scheme for the mechanism is shown in the figure below.[12]
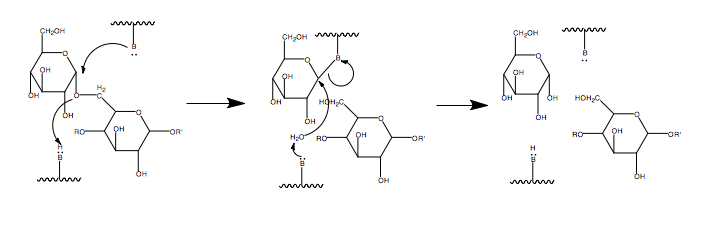
Thus the debranching enzymes, transferase and α-1,6- glucosidase converts the branched glycogen structure into a linear one, paving the way for further cleavage by phosphorylase.
Structure and activity
Two enzymes
In E. coli and other bacteria, glucosyltransferase and glucosidase functions are performed by two distinct enzymes. In E. coli, Glucose transfer is performed by 4-alpha-glucanotransferase, a 78.5 kDa protein coded for by the gene malQ.[13] A second protein, referred to as debranching enzyme, performs α-1,6-glucose cleavage. This enzyme has a molecular mass of 73.6 kDa, and is coded for by the gene glgX.[14] Activity of the two enzymes is not always necessarily coupled. In E. coli glgX selectively catalyzes the cleavage of 4-subunit branches, without the action of glucanotransferase. The product of this cleavage, maltotetraose, is further degraded by maltodextrin phosphorylase.[6][15]
E. coli GlgX is structurally similar to the protein isoamylase. The monomeric protein contains a central domain in which eight parallel beta-strands are surrounded by eight parallel alpha strands. Notable within this structure is a groove 26 angstroms long and 9 angstroms wide, containing aromatic residues that are thought to stabilize a four-glucose branch before cleavage.[6]
The glycogen-degrading enzyme of the archaea Sulfolobus solfataricus, treX, provides an interesting example of using a single active site for two activities: amylosidase and glucanotransferase activities. TreX is structurally similar to glgX, and has a mass of 80kD and one active site.[8][16] Unlike either glgX, however, treX exists as a dimer and tetramer in solution. TreX's oligomeric form seems to play a significant role in altering both enzyme shape and function. Dimerization is thought to stabilize a "flexible loop" located close to the active site. This may be key to explaining why treX (and not glgX) shows glucosyltransferase activity. As a tetramer, the catalytic efficiency of treX is increased fourfold over its dimeric form.[6][17]
One enzyme with two catalytic sites
In mammals and yeast, a single enzyme performs both debranching functions.[18] The human glycogen debranching enzyme (gene: AGL) is a monomer with a molecular weight of 175 kDa. It has been shown that the two catalytic actions of AGL can function independently of each other, demonstrating that multiple active sites are present. This idea has been reinforced with inhibitors of the active site, such as polyhydroxyamine, which were found to inhibit glucosidase activity while transferase activity was not measurably changed.[19] Glycogen debranching enzyme is the only known eukaryotic enzyme that contains multiple catalytic sites and is active as a monomer.[20][21]
Some studies have shown that the C-terminal half of yeast GDE is associated with glucosidase activity, while the N-terminal half is associated with glucosyltransferase activity.[18] In addition to these two active sites, AGL appears to contain a third active site that allows it to bind to a glycogen polymer.[22] It is thought to bind to six glucose molecules of the chain as well as the branched glucose, thus corresponding to 7 subunits within the active site, as shown in the figure below.[23]
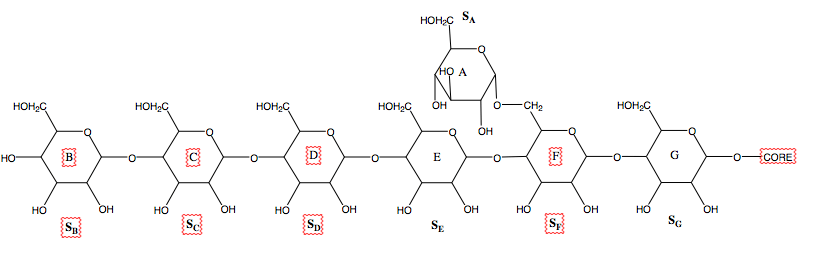
The structure of the Candida glabrata GDE has been reported.[24] The structure revealed that distinct domains in GDE encode the glucanotransferase and glucosidase activities. Their catalyses are similar to that of alpha-amylase and glucoamylase, respectively. Their active sites are selective towards the respective substrates, ensuring proper activation of GDE. Besides the active sites GDE have additional binding sites for glycogen, which are important for its recruitment to glycogen. Mapping the disease-causing mutations onto the GDE structure provided insights into glycogen storage disease type III.
Genetic location
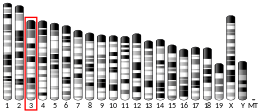
The official name for the gene is "amylo- α- 1,6- glucosidase, 4- α- glucanotransferase", with the official symbol AGL. AGL is an autosomal gene found on chromosome lp21.[10] The AGL gene provides instructions for making several different versions, known as isoforms, of the glycogen debranching enzyme. These isoforms vary by size and are expressed in different tissues, such as liver and muscle. This gene has been studied in great detail, because mutation at this gene is the cause of Glycogen Storage Disease Type III.[25] The gene is 85 kb long, has 35 exons and encodes for a 7.0 kb mRNA. Translation of the gene begins at exon 3,which encodes for the first 27 amino acids of the AGL gene, because the first two exons (68kb) contain the 5' untranslated region. Exons 4-35 encode the remaining 1505 amino acids of the AGL gene.[7] Studies produced by the department of pediatrics at Duke University suggest that the human AGL gene contains at minimum 2 promotor regions, sites where the transcription of the gene begins, that result in differential expression of isoform, different forms of the same protein, mRNAs in a manner that is specific for different tissues.[22][26]
Clinical significance
When GDE activity is compromised, the body cannot effectively release stored glycogen, type III Glycogen Storage Disease (debrancher deficiency), an autosomal recessive disorder, can result. In GSD III glycogen breakdown is incomplete and there is accumulation of abnormal glycogen with short outer branches.[27]
Most patients exhibit GDE defiency in both liver and muscle (Type IIIa), although 15% of patients have retained GDE in muscle while having it absent from the liver (Type IIIb).[10] Depending on mutation location, different mutations in the AGL gene can affect different isoforms of the gene expression. For example, mutations that occur on exon 3, affect the form which affect the isoform that is primarily expressed in the liver; this would lead to GSD type III.[28]
These different manifestation produce varied symptoms, which can be nearly indistinguishable from Type I GSD, including hepatomegaly, hypoglycemia in children, short stature, myopathy, and cardiomyopathy.[7][29] Type IIIa patients often exhibit symptoms related to liver disease and progressive muscle involvement, with variations caused by age of onset, rate of disease progression and severity. Patients with Type IIIb generally symptoms related to liver disease.[30] Type III patients be distinguished by elevated liver enzymes, with normal uric acid and blood lactate levels, differing from other forms of GSD.[28] In patients with muscle involvement, Type IIIa, the muscle weakness becomes predominant into adulthood and can lead to ventricular hypertrophy and distal muscle wasting.[28]
References
- GRCh38: Ensembl release 89: ENSG00000162688 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000033400 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Hers HG, Verhue W, Van hoof F (October 1967). "The determination of amylo-1,6-glucosidase". Eur. J. Biochem. 2 (3): 257–64. doi:10.1111/j.1432-1033.1967.tb00133.x. PMID 6078537.
- Song HN, Jung TY, Park JT, Park BC, Myung PK, Boos W, Woo EJ, Park KH (June 2010). "Structural rationale for the short branched substrate specificity of the glycogen debranching enzyme GlgX". Proteins. 78 (8): 1847–55. doi:10.1002/prot.22697. PMID 20187119.
- Bao Y, Dawson TL, Chen YT (December 1996). "Human glycogen debranching enzyme gene (AGL): complete structural organization and characterization of the 5' flanking region". Genomics. 38 (2): 155–65. doi:10.1006/geno.1996.0611. PMID 8954797.
- Woo EJ, Lee S, Cha H, Park JT, Yoon SM, Song HN, Park KH (October 2008). "Structural insight into the bifunctional mechanism of the glycogen-debranching enzyme TreX from the archaeon Sulfolobus solfataricus". J. Biol. Chem. 283 (42): 28641–8. doi:10.1074/jbc.M802560200. PMC 2661413. PMID 18703518.
- Stryer L, Berg JM, Tymoczko JL (2007). Biochemistry (6th ed.). San Francisco: W.H. Freeman. ISBN 978-0-7167-8724-2.
- Hondoh H, Saburi W, Mori H, et al. (May 2008). "Substrate recognition mechanism of alpha-1,6-glucosidic linkage hydrolyzing enzyme, dextran glucosidase from Streptococcus mutans". J. Mol. Biol. 378 (4): 913–22. doi:10.1016/j.jmb.2008.03.016. PMID 18395742.
- Chiba S (August 1997). "Molecular mechanism in alpha-glucosidase and glucoamylase". Biosci. Biotechnol. Biochem. 61 (8): 1233–9. doi:10.1271/bbb.61.1233. PMID 9301101.
- McCarter JD, Withers SG (December 1994). "Mechanisms of enzymatic glycoside hydrolysis". Curr. Opin. Struct. Biol. 4 (6): 885–92. doi:10.1016/0959-440X(94)90271-2. PMID 7712292.
- "4-alpha-glucanotransferase - Escherichia coli (strain K12)".
- "Glycogen debranching enzyme - Escherichia coli O139:H28 (strain E24377A / ETEC)". UniProt.
- Dauvillée D, Kinderf IS, Li Z, Kosar-Hashemi B, Samuel MS, Rampling L, Ball S, Morell MK (February 2005). "Role of the Escherichia coli glgX gene in glycogen metabolism". J. Bacteriol. 187 (4): 1465–73. doi:10.1128/JB.187.4.1465-1473.2005. PMC 545640. PMID 15687211.
- "TreX - Actinoplanes sp. SN223/29". UniProt.
- Park JT, Park HS, Kang HK, Hong JS, Cha H, Woo EJ, Kim JW, Kim MJ, Boos W, Lee S, Park KH (2008). "Oligomeric and functional properties of a debranching enzyme (TreX) from the archaeon Sulfobus solfataricus P2". Biocatalysis and Biotransformation. 26 (1–2): 76–85. doi:10.1080/10242420701806652.
- Nakayama A, Yamamoto K, Tabata S (August 2001). "Identification of the catalytic residues of bifunctional glycogen debranching enzyme". J. Biol. Chem. 276 (31): 28824–8. doi:10.1074/jbc.M102192200. PMID 11375985.
- Gillard BK, White RC, Zingaro RA, Nelson TE (September 1980). "Amylo-1,6-glucosidase/4-alpha-glucanotransferase. Reaction of rabbit muscle debranching enzyme with an active site-directed irreversible inhibitor, 1-S-dimethylarsino-1-thio-beta-D-glucopyranoside". J. Biol. Chem. 255 (18): 8451–7. PMID 6447697.
- Chen YT, He JK, Ding JH, Brown BI (December 1987). "Glycogen debranching enzyme: purification, antibody characterization, and immunoblot analyses of type III glycogen storage disease". Am. J. Hum. Genet. 41 (6): 1002–15. PMC 1684360. PMID 2961257.
- "Glycogen debranching enzyme - Homo sapiens (Human)". UniProt.
- Gillard BK, Nelson TE (September 1977). "Amylo-1,6-glucosidase/4-alpha-glucanotransferase: use of reversible substrate model inhibitors to study the binding and active sites of rabbit muscle debranching enzyme". Biochemistry. 16 (18): 3978–87. doi:10.1021/bi00637a007. PMID 269742.
- Yamamoto E, Makino Y, Omichi K (May 2007). "Active site mapping of amylo-alpha-1,6-glucosidase in porcine liver glycogen debranching enzyme using fluorogenic 6-O-alpha-glucosyl-maltooligosaccharides". J. Biochem. 141 (5): 627–34. doi:10.1093/jb/mvm065. PMID 17317688.
- Zhai, Liting; Feng, Lingling; Xia, Lin; Yin, Huiyong; Xiang, Song (2016-04-18). "Crystal structure of glycogen debranching enzyme and insights into its catalysis and disease-causing mutations". Nature Communications. 7: ncomms11229. doi:10.1038/ncomms11229. PMC 4837477. PMID 27088557.
- "Genes (Genetic Home Reference a service of U.S. National Library of Medicine". Retrieved February 29, 2012.
- Ding JH, de Barsy T, Brown BI, Coleman RA, Chen YT (January 1990). "Immunoblot analyses of glycogen debranching enzyme in different subtypes of glycogen storage disease type III". J. Pediatr. 116 (1): 95–100. doi:10.1016/S0022-3476(05)81652-X. PMID 2295969.
- Monga SP (2010). Molecular Pathology of Liver Diseases (Molecular Pathology Library). Berlin: Springer. ISBN 978-1-4419-7106-7.
- Shen J, Bao Y, Liu HM, Lee P, Leonard JV, Chen YT (July 1996). "Mutations in exon 3 of the glycogen debranching enzyme gene are associated with glycogen storage disease type III that is differentially expressed in liver and muscle". J. Clin. Invest. 98 (2): 352–7. doi:10.1172/JCI118799. PMC 507437. PMID 8755644.
- Talente GM, Coleman RA, Alter C, Baker L, Brown BI, Cannon RA, et al. (February 1994). "Glycogen storage disease in adults". Ann. Intern. Med. 120 (3): 218–26. doi:10.7326/0003-4819-120-3-199402010-00008. PMID 8273986.
- Kishnani PS, Austin SL, Arn P, Bali DS, Boney A, Case LE, et al. (July 2010). "Glycogen storage disease type III diagnosis and management guidelines". Genetics in Medicine. 12 (7): 446–63. doi:10.1097/GIM.0b013e3181e655b6. PMID 20631546.
External links
| Wikimedia Commons has media related to Glycogen debranching enzyme. |
- GeneReviews/NCBI/NIH/UW entry on Glycogen Storage Disease Type III
- OMIM entries on Glycogen Storage Disease Type III
- Glycogen+debranching+enzyme at the US National Library of Medicine Medical Subject Headings (MeSH)