Sphingolipidoses
Sphingolipidoses are a class of lipid storage disorders relating to sphingolipid metabolism. The main members of this group are Niemann–Pick disease, Fabry disease, Krabbe disease, Gaucher disease, Tay–Sachs disease and metachromatic leukodystrophy. They are generally inherited in an autosomal recessive fashion, but notably Fabry disease is X-linked recessive. Taken together, sphingolipidoses have an incidence of approximately 1 in 10,000, but substantially more in certain populations such as Ashkenazi Jews. Enzyme replacement therapy is available to treat mainly Fabry disease and Gaucher disease, and people with these types of sphingolipidoses may live well into adulthood. The other types are generally fatal by age 1 to 5 years for infantile forms, but progression may be mild for juvenile- or adult-onset forms.
| Sphingolipidoses | |
|---|---|
| Other names | Sphingolipidosis |
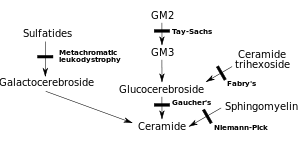 | |
| Diagram showing some of the sphingolipidoses | |
| Specialty | Medical genetics |
Accumulated products
Comparison
| Disease | Deficient enzyme[1] | Accumulated products[1] | Symptoms[1] | Inheritance[1] | Incidence | Generally accepted treatments | Prognosis |
|---|---|---|---|---|---|---|---|
| Niemann-Pick disease | Sphingomyelinase | Sphingomyelin in brain and RBCs |
|
Autosomal recessive | 1 in 100,000[2] | Limited | Usually fatal by the age of approx 1.5 years but may live well into adulthood[3] |
| Fabry disease | α-galactosidase A | Glycolipids, particularly ceramide trihexoside, in brain, heart, kidney |
|
X-linked[4] | Between 1 in 40,000 to 1 in 120,000 live births for males[5] | Enzyme replacement therapy (but expensive) | Life expectancy among males of approximately 60 years[6] |
| Krabbe disease | Galactocerebrosidase | Glycolipids, particularly galactocerebroside, in oligodendrocytes |
|
Autosomal recessive | About 1 in 100,000 births[7] | Bone marrow transplant (high risk, potential failure, effectively provides enzyme replacement to the central nervous system from six months post-transplant, if done in the earliest stages; less effective enzyme replacement provision for the peripheral nervous system) | Untransplanted, and in the case of a failed transplant, generally fatal before age 2 for infants |
| Gaucher disease | Glucocerebrosidase | Glucocerebrosides in RBCs, liver and spleen |
|
Autosomal recessive | About 1 in 20,000 live births,[8] more among Ashkenazi Jews | Enzyme replacement therapy (but expensive) | May live well into adulthood |
| Tay–Sachs disease | Hexosaminidase A | GM2 gangliosides in neurons |
|
Autosomal recessive | Approximately 1 in 320,000 newborns in the general population,[9] more in Ashkenazi Jews | None | Death by approx. 4 years for infantile Tay–Sachs[10] |
| Metachromatic leukodystrophy (MLD) | Arylsulfatase A or prosaposin | Sulfatide compounds in neural tissue | Demyelination in CNS and PNS:
|
Autosomal recessive[11] | 1 in 40,000 to 1 in 160,000[12] | Bone marrow transplant (high risk, potential failure, effectively provides enzyme replacement to the central nervous system from six months post-transplant, if done in the earliest stages; less effective enzyme replacement provision for the peripheral nervous system) | Untransplanted, and in the case of a failed transplant, death by approx. 5 years for infantile MLD |
Metabolic pathways
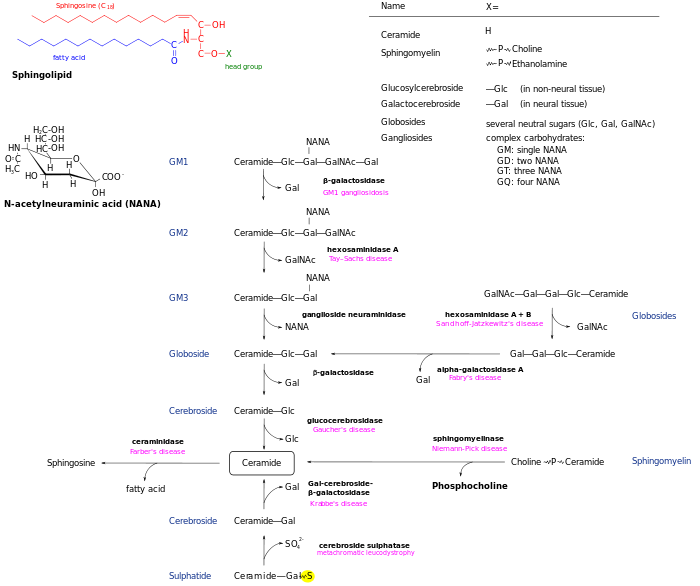
See also
References
- If not otherwise specified, reference is: Marks, Dawn B.; Swanson, Todd; Sandra I Kim; Marc Glucksman (2007). Biochemistry and molecular biology. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins. ISBN 0-7817-8624-X.
- Niemann-Pick disease from Genetics Home Reference. Reviewed: January 2008. Based on an incidence in a general population of 1 in 250,000 for types A and B and 1 in 150,000 for type C
- NINDS Niemann-Pick Disease Information Page at the National Institute of Neurological Disorders and Stroke. Last updated October 6, 2011
- Banikazemi M, Desnick RJ, Astrin KH (2009-07-08). "Fabry Disease". eMedicine Pediatrics: Genetics and Metabolic Disease. Medscape. Retrieved 2010-12-31.
- Mehta, A.; Ricci, R.; Widmer, U.; Dehout, F.; Garcia De Lorenzo, A.; Kampmann, C.; Linhart, A.; Sunder-Plassmann, G.; Ries, M.; Beck, M. (2004). "Fabry disease defined: Baseline clinical manifestations of 366 patients in the Fabry Outcome Survey". European Journal of Clinical Investigation. 34 (3): 236–242. doi:10.1111/j.1365-2362.2004.01309.x. PMID 15025684.
- Waldek, S.; Patel, M. R.; Banikazemi, M.; Lemay, R.; Lee, P. (2009). "Life expectancy and cause of death in males and females with Fabry disease: Findings from the Fabry Registry". Genetics in Medicine. 11 (11): 790–796. doi:10.1097/GIM.0b013e3181bb05bb. PMID 19745746.
- "Krabbe disease". Genetics Home Reference. United States National Library of Medicine. 2008-05-02. Retrieved 2008-05-07.
- Gaucher Disease at National Gaucher Foundation. Retrieved June 2012
- GM2 Gangliosidoses – Introduction And Epidemiology at Medscape. Author: David H Tegay. Updated: Mar 9, 2012
- Colaianni, Alessandra; Chandrasekharan, Subhashini; Cook-Deegan, Robert (2010). "Impact of Gene Patents and Licensing Practices on Access to Genetic Testing and Carrier Screening for Tay–Sachs and Canavan Disease". Genetics in Medicine. 12 (4 Suppl): S5–S14. doi:10.1097/GIM.0b013e3181d5a669. PMC 3042321. PMID 20393311.
- Gieselmann V, Zlotogora J, Harris A, Wenger DA, Morris CP (1994). "Molecular genetics of metachromatic leukodystrophy". Hum. Mutat. 4 (4): 233–42. doi:10.1002/humu.1380040402. PMID 7866401.
- Metachromatic leukodystrophy at Genetics Home Reference. Reviewed September 2007
External links
| Classification |
|---|
- Sphingolipidoses at the US National Library of Medicine Medical Subject Headings (MeSH)