Nuclear reprocessing
Nuclear reprocessing is the chemical separation of fission products and unused uranium from spent nuclear fuel.[1] Originally, reprocessing was used solely to extract plutonium for producing nuclear weapons. With commercialization of nuclear power, the reprocessed plutonium was recycled back into MOX nuclear fuel for thermal reactors.[2] The reprocessed uranium, also known as the spent fuel material, can in principle also be re-used as fuel, but that is only economical when uranium supply is low and prices are high. A breeder reactor is not restricted to using recycled plutonium and uranium. It can employ all the actinides, closing the nuclear fuel cycle and potentially multiplying the energy extracted from natural uranium by about 60 times.[3][4]
Reprocessing must be highly controlled and carefully executed in advanced facilities by highly specialized personnel. Fuel bundles which arrive at the sites from nuclear power plants (after having cooled down for several years) are completely dissolved in chemical baths, which could pose contamination risks if not properly managed. Thus, a reprocessing factory must be considered an advanced chemical site, rather than a nuclear one.
Relatively high cost is associated with spent fuel reprocessing compared to the once-through fuel cycle, but fuel utilization can be increased and waste volumes decreased.[5] Nuclear fuel reprocessing is performed routinely in Europe, Russia and Japan. In the United States, the Obama administration stepped back from President Bush's plans for commercial-scale reprocessing and reverted to a program focused on reprocessing-related scientific research.[6]
Separated components and disposition
The potentially useful components dealt with in nuclear reprocessing comprise specific actinides (plutonium, uranium, and some minor actinides). The lighter elements components include fission products, activation products, and cladding.
| material | disposition |
|---|---|
| plutonium, minor actinides, reprocessed uranium | fission in fast, fusion, or subcritical reactor |
| reprocessed uranium, cladding, filters | less stringent storage as intermediate-level waste |
| long-lived fission and activation products | nuclear transmutation or geological repository |
| medium-lived fission products 137Cs and 90Sr | medium-term storage as high-level waste |
| useful radionuclides and noble metals | industrial and medical uses |
History
The first large-scale nuclear reactors were built during World War II. These reactors were designed for the production of plutonium for use in nuclear weapons. The only reprocessing required, therefore, was the extraction of the plutonium (free of fission-product contamination) from the spent natural uranium fuel. In 1943, several methods were proposed for separating the relatively small quantity of plutonium from the uranium and fission products. The first method selected, a precipitation process called the bismuth phosphate process, was developed and tested at the Oak Ridge National Laboratory (ORNL) between 1943 and 1945 to produce quantities of plutonium for evaluation and use in the US weapons programs. ORNL produced the first macroscopic quantities (grams) of separated plutonium with these processes.
The bismuth phosphate process was first operated on a large scale at the Hanford Site, in the later part of 1944. It was successful for plutonium separation in the emergency situation existing then, but it had a significant weakness: the inability to recover uranium.
The first successful solvent extraction process for the recovery of pure uranium and plutonium was developed at ORNL in 1949. The PUREX process is the current method of extraction. Separation plants were also constructed at Savannah River Site and a smaller plant at West Valley Reprocessing Plant which closed by 1972 because of its inability to meet new regulatory requirements.[7]
Reprocessing of civilian fuel has long been employed at the COGEMA La Hague site in France, the Sellafield site in the United Kingdom, the Mayak Chemical Combine in Russia, and at sites such as the Tokai plant in Japan, the Tarapur plant in India, and briefly at the West Valley Reprocessing Plant in the United States.
In October 1976,[8] concern of nuclear weapons proliferation (especially after India demonstrated nuclear weapons capabilities using reprocessing technology) led President Gerald Ford to issue a Presidential directive to indefinitely suspend the commercial reprocessing and recycling of plutonium in the U.S. On 7 April 1977, President Jimmy Carter banned the reprocessing of commercial reactor spent nuclear fuel. The key issue driving this policy was the risk of nuclear weapons proliferation by diversion of plutonium from the civilian fuel cycle, and to encourage other nations to follow the USA lead.[9][10][11] After that, only countries that already had large investments in reprocessing infrastructure continued to reprocess spent nuclear fuel. President Reagan lifted the ban in 1981, but did not provide the substantial subsidy that would have been necessary to start up commercial reprocessing.[12]
In March 1999, the U.S. Department of Energy (DOE) reversed its policy and signed a contract with a consortium of Duke Energy, COGEMA, and Stone & Webster (DCS) to design and operate a mixed oxide (MOX) fuel fabrication facility. Site preparation at the Savannah River Site (South Carolina) began in October 2005.[13] In 2011 the New York Times reported "...11 years after the government awarded a construction contract, the cost of the project has soared to nearly $5 billion. The vast concrete and steel structure is a half-finished hulk, and the government has yet to find a single customer, despite offers of lucrative subsidies." TVA (currently the most likely customer) said in April 2011 that it would delay a decision until it could see how MOX fuel performed in the nuclear accident at Fukushima Daiichi.[14]
Separation technologies
Water and organic solvents
PUREX
PUREX, the current standard method, is an acronym standing for Plutonium and Uranium Recovery by EXtraction. The PUREX process is a liquid-liquid extraction method used to reprocess spent nuclear fuel, to extract uranium and plutonium, independent of each other, from the fission products. This is the most developed and widely used process in the industry at present.
When used on fuel from commercial power reactors the plutonium extracted typically contains too much Pu-240 to be considered "weapons-grade" plutonium, ideal for use in a nuclear weapon. Nevertheless, highly reliable nuclear weapons can be built at all levels of technical sophistication using reactor-grade plutonium.[15] Moreover, reactors that are capable of refueling frequently can be used to produce weapon-grade plutonium, which can later be recovered using PUREX. Because of this, PUREX chemicals are monitored.[16]
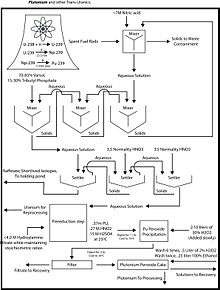
Modifications of PUREX
UREX
The PUREX process can be modified to make a UREX (URanium EXtraction) process which could be used to save space inside high level nuclear waste disposal sites, such as the Yucca Mountain nuclear waste repository, by removing the uranium which makes up the vast majority of the mass and volume of used fuel and recycling it as reprocessed uranium.
The UREX process is a PUREX process which has been modified to prevent the plutonium from being extracted. This can be done by adding a plutonium reductant before the first metal extraction step. In the UREX process, ~99.9% of the uranium and >95% of technetium are separated from each other and the other fission products and actinides. The key is the addition of acetohydroxamic acid (AHA) to the extraction and scrub sections of the process. The addition of AHA greatly diminishes the extractability of plutonium and neptunium, providing somewhat greater proliferation resistance than with the plutonium extraction stage of the PUREX process.
TRUEX
Adding a second extraction agent, octyl(phenyl)-N, N-dibutyl carbamoylmethyl phosphine oxide(CMPO) in combination with tributylphosphate, (TBP), the PUREX process can be turned into the TRUEX (TRansUranic EXtraction) process. TRUEX was invented in the USA by Argonne National Laboratory and is designed to remove the transuranic metals (Am/Cm) from waste. The idea is that by lowering the alpha activity of the waste, the majority of the waste can then be disposed of with greater ease. In common with PUREX this process operates by a solvation mechanism.
DIAMEX
As an alternative to TRUEX, an extraction process using a malondiamide has been devised. The DIAMEX (DIAMide EXtraction) process has the advantage of avoiding the formation of organic waste which contains elements other than carbon, hydrogen, nitrogen, and oxygen. Such an organic waste can be burned without the formation of acidic gases which could contribute to acid rain (although the acidic gases could be recovered by a scrubber). The DIAMEX process is being worked on in Europe by the French CEA. The process is sufficiently mature that an industrial plant could be constructed with the existing knowledge of the process.[17] In common with PUREX this process operates by a solvation mechanism.
SANEX
Selective ActiNide EXtraction. As part of the management of minor actinides it has been proposed that the lanthanides and trivalent minor actinides should be removed from the PUREX raffinate by a process such as DIAMEX or TRUEX. In order to allow the actinides such as americium to be either reused in industrial sources or used as fuel, the lanthanides must be removed. The lanthanides have large neutron cross sections and hence they would poison a neutron driven nuclear reaction. To date the extraction system for the SANEX process has not been defined, but currently several different research groups are working towards a process. For instance the French CEA is working on a bis-triazinyl pyridine (BTP) based process.[18][19][20] Other systems such as the dithiophosphinic acids are being worked on by some other workers.
UNEX
The UNiversal EXtraction process was developed in Russia and the Czech Republic; it is designed to completely remove the most troublesome radioisotopes (Sr, Cs and minor actinides) from the raffinate remaining after the extraction of uranium and plutonium from used nuclear fuel.[21][22] The chemistry is based upon the interaction of caesium and strontium with polyethylene glycol[23][24] and a cobalt carborane anion (known as chlorinated cobalt dicarbollide). The actinides are extracted by CMPO, and the diluent is a polar aromatic such as nitrobenzene. Other dilents such as meta-nitrobenzotrifluoride and phenyl trifluoromethyl sulfone[25] have been suggested as well.
Electrochemical and Ion Exchange methods
An exotic method using electrochemistry and ion exchange in ammonium carbonate has been reported.[26] Other methods for the extraction of uranium using ion exchange in alkaline carbonate and “fumed” lead oxide have also been reported. [27]
Obsolete methods
Bismuth phosphate
The bismuth phosphate process is an obsolete process that adds significant unnecessary material to the final radioactive waste. The bismuth phosphate process has been replaced by solvent extraction processes. The bismuth phosphate process was designed to extract plutonium from aluminium-clad nuclear fuel rods, containing uranium. The fuel was decladded by boiling it in caustic soda. After decladding, the uranium metal was dissolved in nitric acid.
The plutonium at this point is in the +4 oxidation state. It was then precipitated out of the solution by the addition of bismuth nitrate and phosphoric acid to form the bismuth phosphate. The plutonium was coprecipitated with this. The supernatant liquid (containing many of the fission products) was separated from the solid. The precipitate was then dissolved in nitric acid before the addition of an oxidant (such as potassium permanganate) to produce PuO22+. The plutonium was maintained in the +6 oxidation state by addition of a dichromate salt.
The bismuth phosphate was next re-precipitated, leaving the plutonium in solution, and an iron(II) salt (such as ferrous sulfate) was added. The plutonium was again re-precipitated using a bismuth phosphate carrier and a combination of lanthanum salts and fluoride added, forming a solid lanthanum fluoride carrier for the plutonium. Addition of an alkali produced an oxide. The combined lanthanum plutonium oxide was collected and extracted with nitric acid to form plutonium nitrate.[28]
Hexone or redox
This is a liquid-liquid extraction process which uses methyl isobutyl ketone as the extractant. The extraction is by a solvation mechanism. This process has the disadvantage of requiring the use of a salting-out reagent (aluminium nitrate) to increase the nitrate concentration in the aqueous phase to obtain a reasonable distribution ratio (D value). Also, hexone is degraded by concentrated nitric acid. This process has been replaced by the PUREX process.[29][30]
Pu4+ + 4 NO3− + 2 S → [Pu(NO3)4S2]
Butex, β,β'-dibutyoxydiethyl ether
A process based on a solvation extraction process using the triether extractant named above. This process has the disadvantage of requiring the use of a salting-out reagent (aluminium nitrate) to increase the nitrate concentration in the aqueous phase to obtain a reasonable distribution ratio. This process was used at Windscale many years ago. This process has been replaced by PUREX.
Pyroprocessing
Pyroprocessing is a generic term for high-temperature methods. Solvents are molten salts (e.g. LiCl + KCl or LiF + CaF2) and molten metals (e.g. cadmium, bismuth, magnesium) rather than water and organic compounds. Electrorefining, distillation, and solvent-solvent extraction are common steps.
These processes are not currently in significant use worldwide, but they have been pioneered at Argonne National Laboratory[31][32] with current research also taking place at CRIEPI in Japan, the Nuclear Research Institute of Řež in Czech Republic, Indira Gandhi Centre for Atomic Research in India and KAERI in South Korea.[33][34][35][36]
Advantages
- The principles behind them are well understood, and no significant technical barriers exist to their adoption.[37]
- Readily applied to high-burnup spent fuel and requires little cooling time, since the operating temperatures are high already.
- Does not use solvents containing hydrogen and carbon, which are neutron moderators creating risk of criticality accidents and can absorb the fission product tritium and the activation product carbon-14 in dilute solutions that cannot be separated later.
- Alternatively, voloxidation[38] can remove 99% of the tritium from used fuel and recover it in the form of a strong solution suitable for use as a supply of tritium.
- More compact than aqueous methods, allowing on-site reprocessing at the reactor site, which avoids transportation of spent fuel and its security issues, instead storing a much smaller volume of fission products on site as high-level waste until decommissioning. For example, the Integral Fast Reactor and Molten Salt Reactor fuel cycles are based on on-site pyroprocessing.
- It can separate many or even all actinides at once and produce highly radioactive fuel which is harder to manipulate for theft or making nuclear weapons. (However, the difficulty has been questioned.[39]) In contrast the PUREX process was designed to separate plutonium only for weapons, and it also leaves the minor actinides (americium and curium) behind, producing waste with more long-lived radioactivity.
- Most of the radioactivity in roughly 102 to 105 years after the use of the nuclear fuel is produced by the actinides, since there are no fission products with half-lives in this range. These actinides can fuel fast reactors, so extracting and reusing (fissioning) them increases energy production per kg of fuel, as well as reducing the long-term radioactivity of the wastes.
Disadvantages
- Reprocessing as a whole is not currently (2005) in favor, and places that do reprocess already have PUREX plants constructed. Consequently, there is little demand for new pyrometalurgical systems, although there could be if the Generation IV reactor programs become reality.
- The used salt from pyroprocessing is less suitable for conversion into glass than the waste materials produced by the PUREX process.
- If the goal is to reduce the longevity of spent nuclear fuel in burner reactors, then better recovery rates of the minor actinides need to be achieved.
Electrolysis
The electrolysis methods are based on the difference in the standard potentials of uranium, plutonium and minor actinides in a molten salt. The standard potential of uranium is the lowest, therefore when a potential is applied, the uranium will be reduced at the cathode out of the molten salt solution before the other elements.[40]

PYRO-A and -B for IFR
These processes were developed by Argonne National Laboratory and used in the Integral Fast Reactor project.
PYRO-A is a means of separating actinides (elements within the actinide family, generally heavier than U-235) from non-actinides. The spent fuel is placed in an anode basket which is immersed in a molten salt electrolyte. An electric current is applied, causing the uranium metal (or sometimes oxide, depending on the spent fuel) to plate out on a solid metal cathode while the other actinides (and the rare earths) can be absorbed into a liquid cadmium cathode. Many of the fission products (such as caesium, zirconium and strontium) remain in the salt.[41][42][43] As alternatives to the molten cadmium electrode it is possible to use a molten bismuth cathode, or a solid aluminium cathode.[44]
As an alternative to electrowinning, the wanted metal can be isolated by using a molten alloy of an electropositive metal and a less reactive metal.[45]
Since the majority of the long term radioactivity, and volume, of spent fuel comes from actinides, removing the actinides produces waste that is more compact, and not nearly as dangerous over the long term. The radioactivity of this waste will then drop to the level of various naturally occurring minerals and ores within a few hundred, rather than thousands of, years.[46]
The mixed actinides produced by pyrometallic processing can be used again as nuclear fuel, as they are virtually all either fissile, or fertile, though many of these materials would require a fast breeder reactor in order to be burned efficiently. In a thermal neutron spectrum, the concentrations of several heavy actinides (curium-242 and plutonium-240) can become quite high, creating fuel that is substantially different from the usual uranium or mixed uranium-plutonium oxides (MOX) that most current reactors were designed to use.
Another pyrochemical process, the PYRO-B process, has been developed for the processing and recycling of fuel from a transmuter reactor ( a fast breeder reactor designed to convert transuranic nuclear waste into fission products ). A typical transmuter fuel is free from uranium and contains recovered transuranics in an inert matrix such as metallic zirconium. In the PYRO-B processing of such fuel, an electrorefining step is used to separate the residual transuranic elements from the fission products and recycle the transuranics to the reactor for fissioning. Newly generated technetium and iodine are extracted for incorporation into transmutation targets, and the other fission products are sent to waste.
Voloxidation
Voloxidation (for volumetric oxidation) involves heating oxide fuel with oxygen, sometimes with alternating oxidation and reduction, or alternating oxidation by ozone to uranium trioxide with decomposition by heating back to triuranium octoxide.[38] A major purpose is to capture tritium as tritiated water vapor before further processing where it would be difficult to retain the tritium. Other volatile elements leave the fuel and must be recovered, especially iodine, technetium, and carbon-14. Voloxidation also breaks up the fuel or increases its surface area to enhance penetration of reagents in following reprocessing steps.
Volatilization in isolation
Simply heating spent oxide fuel in an inert atmosphere or vacuum at a temperature between 700 °C and 1000 °C as a first reprocessing step can remove several volatile elements, including caesium whose isotope caesium-137 emits about half of the heat produced by the spent fuel over the following 100 years of cooling (however, most of the other half is from strontium-90, which has a similar half-life). The estimated overall mass balance for 20,000 g of processed fuel with 2,000 g of cladding is:[47]
| Input | Residue | Zeolite filter | Carbon filter | Particle filters | |
|---|---|---|---|---|---|
| Palladium | 28 | 14 | 14 | ||
| Tellurium | 10 | 5 | 5 | ||
| Molybdenum | 70 | 70 | |||
| Caesium | 46 | 46 | |||
| Rubidium | 8 | 8 | |||
| Silver | 2 | 2 | |||
| Iodine | 4 | 4 | |||
| Cladding | 2000 | 2000 | |||
| Uranium | 19218 | 19218 | ? | ||
| Others | 614 | 614 | ? | ||
| Total | 22000 | 21851 | 145 | 4 | 0 |
Fluoride volatility
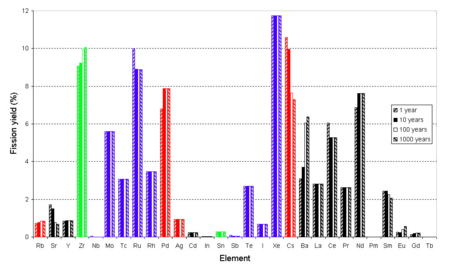
In the fluoride volatility process, fluorine is reacted with the fuel. Fluorine is so much more reactive than even oxygen that small particles of ground oxide fuel will burst into flame when dropped into a chamber full of fluorine. This is known as flame fluorination; the heat produced helps the reaction proceed. Most of the uranium, which makes up the bulk of the fuel, is converted to uranium hexafluoride, the form of uranium used in uranium enrichment, which has a very low boiling point. Technetium, the main long-lived fission product, is also efficiently converted to its volatile hexafluoride. A few other elements also form similarly volatile hexafluorides, pentafluorides, or heptafluorides. The volatile fluorides can be separated from excess fluorine by condensation, then separated from each other by fractional distillation or selective reduction. Uranium hexafluoride and technetium hexafluoride have very similar boiling points and vapor pressures, which makes complete separation more difficult.
Many of the fission products volatilized are the same ones volatilized in non-fluorinated, higher-temperature volatilization, such as iodine, tellurium and molybdenum; notable differences are that technetium is volatilized, but caesium is not.
Some transuranium elements such as plutonium, neptunium and americium can form volatile fluorides, but these compounds are not stable when the fluorine partial pressure is decreased.[48] Most of the plutonium and some of the uranium will initially remain in ash which drops to the bottom of the flame fluorinator. The plutonium-uranium ratio in the ash may even approximate the composition needed for fast neutron reactor fuel. Further fluorination of the ash can remove all the uranium, neptunium, and plutonium as volatile fluorides; however, some other minor actinides may not form volatile fluorides and instead remain with the alkaline fission products. Some noble metals may not form fluorides at all, but remain in metallic form; however ruthenium hexafluoride is relatively stable and volatile.
Distillation of the residue at higher temperatures can separate lower-boiling transition metal fluorides and alkali metal (Cs, Rb) fluorides from higher-boiling lanthanide and alkaline earth metal (Sr, Ba) and yttrium fluorides. The temperatures involved are much higher, but can be lowered somewhat by distilling in a vacuum. If a carrier salt like lithium fluoride or sodium fluoride is being used as a solvent, high-temperature distillation is a way to separate the carrier salt for reuse.
Molten salt reactor designs carry out fluoride volatility reprocessing continuously or at frequent intervals. The goal is to return actinides to the molten fuel mixture for eventual fission, while removing fission products that are neutron poisons, or that can be more securely stored outside the reactor core while awaiting eventual transfer to permanent storage.
Chloride volatility and solubility
Many of the elements that form volatile high-valence fluorides will also form volatile high-valence chlorides. Chlorination and distillation is another possible method for separation. The sequence of separation may differ usefully from the sequence for fluorides; for example, zirconium tetrachloride and tin tetrachloride have relatively low boiling points of 331 °C and 114.1 °C. Chlorination has even been proposed as a method for removing zirconium fuel cladding,[38] instead of mechanical decladding.
Chlorides are likely to be easier than fluorides to later convert back to other compounds, such as oxides.
Chlorides remaining after volatilization may also be separated by solubility in water. Chlorides of alkaline elements like americium, curium, lanthanides, strontium, caesium are more soluble than those of uranium, neptunium, plutonium, and zirconium.
Radioanalytical separations
In order to determine the distribution of radioactive metals for analytical purposes, Solvent Impregnated Resins (SIRs) can be used. SIRs are porous particles, which contain an extractant inside their pores. This approach avoids the liquid-liquid separation step required in conventional liquid-liquid extraction. For the preparation of SIRs for radioanalytical separations, organic Amberlite XAD-4 or XAD-7 can be used. Possible extractants are e.g. trihexyltetradecylphosphonium chloride(CYPHOS IL-101) or N,N0-dialkyl-N,N0-diphenylpyridine-2,6-dicarboxyamides (R-PDA; R = butyl, octy I, decyl, dodecyl).[49]
Economics
The relative economics of reprocessing-waste disposal and interim storage-direct disposal was the focus of much debate over the first decade of the 2000s. Studies[50] have modeled the total fuel cycle costs of a reprocessing-recycling system based on one-time recycling of plutonium in existing thermal reactors (as opposed to the proposed breeder reactor cycle) and compare this to the total costs of an open fuel cycle with direct disposal. The range of results produced by these studies is very wide, but all are agreed that under current (2005) economic conditions the reprocessing-recycle option is the more costly.[51]
If reprocessing is undertaken only to reduce the radioactivity level of spent fuel it should be taken into account that spent nuclear fuel becomes less radioactive over time. After 40 years its radioactivity drops by 99.9%,[52] though it still takes over a thousand years for the level of radioactivity to approach that of natural uranium.[53] However the level of transuranic elements, including plutonium-239, remains high for over 100,000 years, so if not reused as nuclear fuel, then those elements need secure disposal because of nuclear proliferation reasons as well as radiation hazard.
On 25 October 2011 a commission of the Japanese Atomic Energy Commission revealed during a meeting calculations about the costs of recycling nuclear fuel for power generation. These costs could be twice the costs of direct geological disposal of spent fuel: the cost of extracting plutonium and handling spent fuel was estimated at 1.98 to 2.14 yen per kilowatt-hour of electricity generated. Discarding the spent fuel as waste would cost only 1 to 1.35 yen per kilowatt-hour.[54][55]
In July 2004 Japanese newspapers reported that the Japanese Government had estimated the costs of disposing radioactive waste, contradicting claims four months earlier that no such estimates had been made. The cost of non-reprocessing options was estimated to be between a quarter and a third ($5.5–7.9 billion) of the cost of reprocessing ($24.7 billion). At the end of the year 2011 it became clear that Masaya Yasui, who had been director of the Nuclear Power Policy Planning Division in 2004, had instructed his subordinate in April 2004 to conceal the data. The fact that the data were deliberately concealed obliged the ministry to re-investigate the case and to reconsider whether to punish the officials involved.[56][57]
List of sites
| Country | Reprocessing site | Fuel type | Procedure | Reprocessing capacity tHM/yr |
Commissioning or operating period |
|---|---|---|---|---|---|
| Mol | LWR, MTR (Material test reactor) | 80[58] | 1966–1974[58] | ||
| intermediate pilot plant[59] | 60–100 | 1968-early 1970s | |||
| Plant 404[60] | 50 | 2004 | |||
| Karlsruhe, WAK | LWR[61] | 35[58] | 1971–1990[58] | ||
| Marcoule, UP 1 | Military | 1,200[58] | 1958[58]-1997[62] | ||
| Marcoule, CEA APM | FBR | PUREX DIAMEX SANEX[63] | 6[61] | 1988–present[61] | |
| La Hague, UP 2 | LWR[61] | PUREX[64] | 900[58] | 1967–1974[58] | |
| La Hague, UP 2–400 | LWR[61] | PUREX[64] | 400[58] | 1976–1990[58] | |
| La Hague, UP 2–800 | LWR | PUREX[64] | 800[58] | 1990[58] | |
| La Hague, UP 3 | LWR | PUREX[64] | 800[58] | 1990[58] | |
| Windscale | Magnox | BUTEX | 1,000[58] | 1956–1962[58] | |
| Sellafield, B205 | Magnox[61] | PUREX | 1,500[58] | 1964[58] | |
| Dounreay | FBR[61] | 8[58] | 1980[58] | ||
| THORP | AGR, LWR | PUREX | 900[58][65] | 1994[58][65]-2018 | |
| Rotondella | Thorium | 5[58] | 1968[58] (shutdown) | ||
| Trombay | Military | PUREX[66] | 60[58] | 1965[58] | |
| Tarapur | PHWR | PUREX | 100[58] | 1982[58] | |
| Kalpakkam | PHWR and FBTR | PUREX | 100[67] | 1998[67] | |
| Tarapur | PHWR | 100[68] | 2011[68] | ||
| Dimona | Military | 60–100[69] | ~1960–present | ||
| Tokaimura | LWR[70] | 210[58] | 1977[58]-2006[71] | ||
| Rokkasho | LWR[61] | 800[58][65] | 2021[72] | ||
| New Labs, Rawalpindi | Military/Plutonium/Thorium | 80[73] | 1982–present | ||
| Khushab Nuclear Complex, Atomic City of Pakistan | HWR/Military/Tritium | 22 kg[74] | 1986–present | ||
| Mayak Plant B | Military | 400 | 1948-196?[75] | ||
| Mayak Plant BB, RT-1 | LWR[61] | PUREX + Np separation[76] | 400[58][65] | 1978[58] | |
| Tomsk-7 Radiochemical Plant | Military | 6000[69] | 1956[77] | ||
| Zheleznogorsk (Krasnoyarsk-26) | Military | 3500[69] | 1964–~2010[78] | ||
| Zheleznogorsk, RT-2 | VVER | 800[58] | under construction (2030) | ||
| Hanford Site | Military | bismuth phosphate, REDOX, PUREX | 1944–1988[79] | ||
| Savannah River Site | Military/LWR/HWR/Tritium | PUREX, REDOX, THOREX, Np separation | 5000[80] | 1952–2002 | |
| West Valley | LWR[61] | PUREX | 300[58] | 1966–1972[58] | |
| Barnwell | LWR | PUREX | 1500 | never permitted to operate[81] | |
| INL | LWR | PUREX | - |
See also
- Nuclear fuel cycle
- Breeder reactor
- Nuclear fusion-fission hybrid
- Spent nuclear fuel shipping cask
- Taylor Wilson's nuclear waste-fired small reactor
- Global Nuclear Energy Partnership announced February, 2006
References
- Andrews, A. (27 March 2008). Nuclear Fuel Reprocessing: U.S. Policy. CRS Report For Congress. Retrieved 25 March 2011
- MOX fuel can extend the energy extracted by about 12% but slightly reduces plutonium stocks. Information from the World Nuclear Association about MOX
- "Supply of Uranium". World Nuclear Association. Retrieved 29 January 2010.
- "Fast Neutron Reactors". World Nuclear Association. Retrieved 11 March 2012.
- Harold Feiveson; et al. (2011). "Managing nuclear spent fuel: Policy lessons from a 10-country study". Bulletin of the Atomic Scientists.
- "Adieu to nuclear recycling". Nature. 460 (7252): 152. 2009. Bibcode:2009Natur.460R.152.. doi:10.1038/460152b. PMID 19587715.
- "Plutonium Recovery from Spent Fuel Reprocessing by Nuclear Fuel Services at West Valley, New York from 1966 to 1972". U.S. Department of Energy. February 1996. Retrieved 17 June 2007.
- Gerald Ford 28 October 1976 Statement on Nuclear Policy. Retrieved 30 June 2012.
- Dr. Ned Xoubi (2008). "The Politics, Science, Environment, and common sense of Spent Nuclear Fuel Reprocessing 3 decades Later" (PDF). Symposium on the Technology of Peaceful Nuclear Energy, Irbid, Jordan. Archived from the original (PDF) on 16 May 2011.
- "Depleted Cranium » Blog Archive » Why You Can't Build a Bomb from Spent Fuel". Archived from the original on 4 February 2012.
- "Proving a Negative - Why Modern Used Nuclear Fuel Cannot Be Used to Make a Weapon - Atomic Insights". atomicinsights.com. 17 February 2015. Retrieved 4 April 2018.
- Nuclear Fuel Reprocessing: U.S. Policy Development. (PDF). Retrieved 10 December 2011.
- Duke, Cogema, Stone & Webster (DCS) Reports sent to NRC. Nrc.gov. Retrieved 10 December 2011.
- New Doubts About Turning Plutonium Into a Fuel, 10 April 2011
- U.S. Program for Disposition of Excess Weapons Plutonium, IAEA-SM-346/102, Matthew Bunn, 2002.
- Irvine, Maxwell (2011). Nuclear power : a very short introduction. Oxford: Oxford University Press. p. 55. ISBN 9780199584970.
- "Nuclear Energy: Fuel of the Future?". Princeton University. Archived from the original on 1 October 2012. Retrieved 6 April 2013.
- C. Hill, D. Guillaneux, X. Hérès, N. Boubals and L. Ramain SANEX-BTP PROCESS DEVELOPMENT STUDIES Archived 15 November 2012 at the Wayback Machine
- C. Hill, L. Berthon, P. Bros, J-P. Dancausse and D. Guillaneux SANEX-BTP PROCESS DEVELOPMENT STUDIES. Commissariat à l'Énergie Atomique
- Béatrice Rat, Xavier Hérès Modelling and achievement of a SANEX process flowsheet for trivalent actinides/lanthanides separation using BTP extractant (bis-1,2,4-triazinyl-pyridine). Archived 16 October 2005 at the Wayback Machine
- "U.S.-Russia Team Makes Treating Nuclear Waste Easier". U.S. embassy press release(?). 19 December 2001. Archived from the original on 28 July 2014. Retrieved 14 June 2007.
- J. Banaee; et al. (1 September 2001). "INTEC High-Level Waste Studies Universal Solvent Extraction Feasibility Study". INEEL Technical report.
- Law, Jack D.; Herbst, R. Scott; Todd, Terry A.; Romanovskiy, Valeriy N.; Babain, Vasily A.; Esimantovskiy, Vyatcheslav M.; Smirnov, Igor V.; Zaitsev, Boris N. (2001). "The Universal Solvent Extraction (Unex) Process. Ii. Flowsheet Development and Demonstration of the Unex Process for the Separation of Cesium, Strontium, and Actinides from Actual Acidic Radioactive Waste". Solvent Extraction and Ion Exchange. 19: 23. doi:10.1081/SEI-100001371.
- Romanovskiy, Valeriy N.; Smirnov, Igor V.; Babain, Vasily A.; Todd, Terry A.; Herbst, R. Scott; Law, Jack D.; Brewer, Ken N. (2001). "The Universal Solvent Extraction (Unex) Process. I. Development of the Unex Process Solvent for the Separation of Cesium, Strontium, and the Actinides from Acidic Radioactive Waste". Solvent Extraction and Ion Exchange. 19: 1. doi:10.1081/SEI-100001370.
- J.D. Law; et al. (1 March 2001). "Flowsheet testing of the universal solvent extraction process for the simultaneous separation of caesium, strontium, and the actinides from dissolved INEEL calcine" (PDF). WM 2001 conference proceedings. Archived from the original (PDF) on 28 September 2007. Retrieved 17 June 2006.
- Asanuma, Noriko; et al. (2006). "Andodic dissociation of UO2 pellet containing simulated fission products in ammonium carbonate solution". Journal of Nuclear Science and Technology. 43 (3): 255–262. doi:10.3327/jnst.43.255.
- , Gardner, Harry E., "Recovery of Uranium from Uranium Bearing Solutions Containing Molybdenum", issued 1979-12-14
- Gerber, Michelle. "The plutonium production story at the Hanford Site: processes and facilities history (WHC-MR-0521) (excerpts)". Department of Energy.
- Seaborg, Glenn T.; et al. (23 August 1960). "Method for separation of plutonium from uranium and fission products by solvent extraction". U.S. Patent and Trademark Office. OSTI 4134289. Cite journal requires
|journal=(help) U.S. Patent 2,950,166 - L.W. Gray (15 April 1999). "From separations to reconstitution—a short history of plutonium in the U.S. and Russia (UCRL-JC-133802)" (PDF). Lawrence Livermore National Laboratory preprint.
- "Pyroprocessing Development". Argonne National Laboratory. Retrieved 6 June 2016.
- "Pyroprocessing Technologies: Recycling used nuclear fuel for a sustainable energy future" (PDF). Argonne National Laboratory. 2012. p. 7. Archived from the original (PDF) on 19 February 2013. Retrieved 6 June 2016.
- T. Inoue. "An Overview of CRIEPI Pyroprocessing Activities" (PDF).
- Tulackova, R., et al. "Development of Pyrochemical Reprocessing of the Spent Nuclear Fuel and Prospects of Closed Fuel Cycle." Atom Indonesia 33.1 (2007): 47-59.
- Nagarajan, K., et al. "Current status of pyrochemical reprocessing research in India." Nuclear Technology 162.2 (2008): 259-263.
- Lee, Hansoo, et al. "Development of Pyro-processing Technology at KAERI." (2009).
- "PYROPROCESSING PROGRESS AT IDAHO NATIONAL LABORATORY" (PDF). Idaho National Laboratory article. September 2007. Archived from the original (PDF) on 12 June 2011.
- Guillermo D. Del Cul; et al. "Advanced Head-End Processing of Spent Fuel: A Progress Report" (PDF). 2005 ANS annual meeting. Oak Ridge National Laboratory, U.S. DOE. Archived from the original (PDF) on 7 March 2006. Retrieved 3 May 2008.
- "Limited Proliferation-Resistance Benefits from Recycling Unseparated Transuranics and Lanthanides from Light-Water Reactor Spent Fuel" (PDF). p. 4.
- Morss, L. R. The chemistry of the actinide and transactinide elements. Eds. Lester R. Morss, et al. Vol. 1. Dordrecht: Springer, 2006.
- "Development of pyro-process fuel cell technology" (PDF). CRIEPI News. July 2002. Archived from the original (PDF) on 25 February 2009. Retrieved 22 June 2009.
- Masatoshi Iizuka (12 December 2001). "Development of plutonium recovery process by molten salt electrorefining with liquid cadmium cathode" (PDF). Proceedings of the 6th information exchange meeting on actinide and fission product partitioning and transmutation (Madrid, Spain). Retrieved 22 June 2009.
- R. Tulackova (Zvejskova), K. Chuchvalcova Bimova, P. Soucek, F. Lisy Study of Electrochemical Processes for Separation of the Actinides and Lanthanides in Molten Fluoride Media (PPT file). Nuclear Research Institute Rez plc, Czech Republic
- Electrochemical Behaviours of Lanthanide Fluorides in the Electrolysis System with LiF-NaF-KF Salt. (PDF) . Retrieved 10 December 2011.
- Ionic Liquids/Molten Salts and Lanthanides/Actinides Reference List. Merck.de. Retrieved 10 December 2011.
- "Advanced Fuel Cycle Initiative". U.S. Department of Energy. Archived from the original on 10 May 2012. Retrieved 3 May 2008.
- Wolverton, Daren; et al. (11 May 2005). "Removal of caesium from spent nuclear fuel destined for the electrorefiner fuel treatment process" (PDF). University of Idaho (dissertation?). Archived from the original (PDF) on 29 November 2007.
- Neeb, Karl-Heinz (1997). The radiochemistry of nuclear power plants with light water reactors. Walter de Gruyter. ISBN 978-3-11-013242-7.
- Kabay, N.; Cortina, J.L.; Trochimczuk, A.; Streat, M. (2010). "Solvent-impregnated resins (SIRs) – Methods of preparation and their applications". React. Funct. Polym. 70 (8): 484–496. doi:10.1016/j.reactfunctpolym.2010.01.005. hdl:2117/10365.
- "Advanced Fuel Cycle Cost Basis" (PDF). Idaho National Laboratory, United States Department of Energy. Archived from the original (PDF) on 28 November 2011. Retrieved 19 December 2010.
- "Recycled Nuclear Fuel Cost Calculator". www.wise-uranium.org. Retrieved 4 April 2018.
- "Waste Management and Disposal". World Nuclear Association. Retrieved 3 May 2008.
- "Radioactive Wastes: Myths and Realities". World Nuclear Association. June 2006. Retrieved 3 May 2008.
- NHK-world (26 October 2011) Nuclear fuel recycling costs Archived 10 August 2011 at the Wayback Machine
- JAIF (26 October 2011) Nuclear fuel recycling costs
- "Cover-up of estimated costs to dispose of radioactive waste raises serious questions". The Mainichi Daily News. 2 January 2012. Retrieved 8 January 2012.
- Mycle, Schneider. "Japanese mislead about spent fuel reprocessing costs". International Panel on Fissile Materials. Retrieved 8 January 2012.
- "Reprocessing plants, world-wide". European Nuclear Society. Archived from the original on 22 June 2015. Retrieved 29 July 2008.
- Wright, David; Gronlund, Lisbeth (2003). "Estimating China's Production of Plutonium for Weapons" (PDF). Science & Global Security. 11: 61–80. doi:10.1080/08929880309007.
- All Things Nuclear • China and Reprocessing: Separating Fact from Fiction. Allthingsnuclear.org (11 January 2011). Retrieved 10 December 2011.
- "Civil Reprocessing Facilities" (PDF). Princeton University. Retrieved 30 July 2008.
- "Marcoule – Valrho". Global Security. Retrieved 30 July 2008.
- Dominique Warin (2007). "Status of the French Research Program on Partitioning and Transmutation". Journal of Nuclear Science and Technology. 44 (3): 410. doi:10.3327/jnst.44.410.
- "BASSE-NORMANDIE- LOWER NORMANDY, La Hague". France Nucleaire. Archived from the original on 16 July 2011. Retrieved 31 July 2008.
- "Processing of Used Nuclear Fuel". World Nuclear Association. September 2013. Retrieved 5 December 2013.
- "CIRUS and DHRUVA Reactors, Trombay". Global Security. Retrieved 30 July 2008.
- "Kalpakkam Atomic Reprocessing Plant [KARP]". Global Security. Retrieved 30 July 2008.
- PM to dedicate Tarapur nuke reprocessing unit next week. Business-standard.com. Retrieved 10 December 2011.
- Global Fissile Material Report 2010, International Panel on Fissile Materials http://fissilematerials.org/library/gfmr10.pdf
- "Tokai Reprocessing Plant (TRP)". Global Security. Retrieved 30 July 2008.
- Kramer, D. (2012). "Is Japan ready to forgo nuclear reprocessing?". Physics Today. 65 (3): 25–42. Bibcode:2012PhT....65c..25K. doi:10.1063/PT.3.1469.
- "Further delay to completion of Rokkasho facilities". World Nuclear News. 28 December 2017. Retrieved 28 December 2017.
- "Rawalpindi / Nilhore". Federation of American Scientists. Retrieved 8 March 2000. Check date values in:
|accessdate=(help) - "Pakistan's Indigenous Nuclear Reactor Starts Up". The Nation. 13 April 1998.
- "Chelyabinsk-65". Global Security. Retrieved 29 July 2008.
- S. Guardini; et al. (16 June 2003). "Modernization and Enhancement of NMAC at the Mayak RT-1 Plant". INMM. Archived from the original on 28 July 2014. Retrieved 9 August 2008.
- "Tomsk-7 / Seversk". Global Security. Retrieved 1 June 2020.
- "Krasnoyarsk-26 / Zheleznogorsk". Global Security. Retrieved 1 June 2020.
- "T Plant overview". Department of energy. Retrieved 9 April 2011.
- LeVerne Fernandez. "Savannah River Site Canyons—Nimble Behemoths of the Atomic Age (WSRC-MS-2000-00061)" (PDF).
- "West Valley Demonstration Project", Wikipedia, 1 December 2018, retrieved 13 April 2020
Further reading
- Williamson, M.A.; Willit, J.L. (2011). "Pyroprocessing Flowsheets for Recycling Used Nuclear Fuel" (PDF). Nuclear Engineering and Technology. 43 (4): 329–334. doi:10.5516/NET.2011.43.4.329.
- Till, C.E.; Chang, Y.I; Hannum, W.H. (1997). "The integral fast reactor-an overview". Progress in Nuclear Energy. 31 (1–2): 3–11. doi:10.1016/0149-1970(96)00001-7.
- OECD Nuclear Energy Agency, The Economics of the Nuclear Fuel Cycle, Paris, 1994
- I. Hensing and W Schultz, Economic Comparison of Nuclear Fuel Cycle Options, Energiewirtschaftlichen Instituts, Cologne, 1995.
- Cogema, Reprocessing-Recycling: the Industrial Stakes, presentation to the Konrad-Adenauer-Stiftung, Bonn, 9 May 1995.
- OECD Nuclear Energy Agency, Plutonium Fuel: An Assessment, Paris, 1989.
- National Research Council, "Nuclear Wastes: Technologies for Separation and Transmutation", National Academy Press, Washington D.C. 1996.
External links
- Processing of Used Nuclear Fuel, World Nuclear Association
- PUREX Process, European Nuclear Society
- Mixed Oxide Fuel (MOX) – World Nuclear Association
- Disposal Options for Surplus Weapons-Usable Plutonium – Congressional Research Service Report for Congress
- Brief History of Fuel Reprocessing
- Annotated bibliography for reprocessing spent nuclear fuel from the Alsos Digital Library for Nuclear Issues
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||