Kuwajima Taxol total synthesis
The Kuwajima Taxol total synthesis by the group of Isao Kuwajima of the Tokyo Institute of Technology is one of several efforts in taxol total synthesis published in the 1990s.[1][2] The total synthesis of Taxol is considered a landmark in organic synthesis.
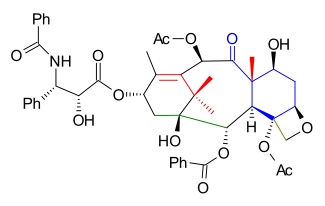
This synthesis is truly synthetic without any help from small biomolecule precursors and also a linear synthesis with molecule ring construction in the order of A, B, C, D. At some point chirality is locked into the molecule via an asymmetric synthesis step which is unique compared to the other efforts. In common with the other efforts the tail addition is based on the Ojima lactam.
The 20 carbon frame is constructed from several pieces: propargyl alcohol (C1, C2, C14), propionaldehyde (C13, C12, C18), isobutyric acid (C15, C16, C17, C11), Trimethyl(phenylthiomethyl)silane (C10), 2-bromobenzaldehyde (C3 to C9), diethylaluminum cyanide (C19) and trimethylsilylmethyl bromide (C20)
Synthesis A ring
Ring A synthesis (scheme 1) started by joining the THP protected propargyl alcohol 1.1 (the C2-C1-C14 fragment) and propionaldehyde 1.2 (fragment C13-C12-C18) in a nucleophilic addition with n-butyllithium to alcohol 1.3. The Lindlar catalyst then reduced the alkyne to the alkene in 1.4 and Swern oxidation converted the alcohol group to the enone group in 1.5. Fragment C11-C15-C16-C17 1.6 was then added as the lithium enolate of isobutyric acid ethyl ester in a conjugate addition to gamma keto ester 1.7. A Claisen condensation closed the ring to 1.8 and the intermediate enol is captured by pivaloyl chloride (piv) as a protective group. The THP group was removed with TsOH to 1.9 and the formed alcohol oxidized by Swern oxidation to aldehyde 1.10. The TIPS silyl enol ether 1.11 was formed by reaction with the triflate TIPSOtf and DBU in DMAP setting the stage for asymmetric dihydroxylation to hydroxyaldehyde 1.12. The piv protecting group was then replaced by a TIPS group in 1.14 after protecting the aldehyde as the aminal 1.13 and as this group is automatically lost on column chromatography, the step was repeated to aminal 1.15. The C10 fragment was then introduced by the lithium salt of Trimethyl(phenylthiomethyl)silane 1.16 in a Peterson olefination to the sulfide 1.17 followed by deprotection to completed ring A 1.18. The A ring is now complete with the aldehyde group and de sulfide group in place for anchoring with ring C forming ring B.
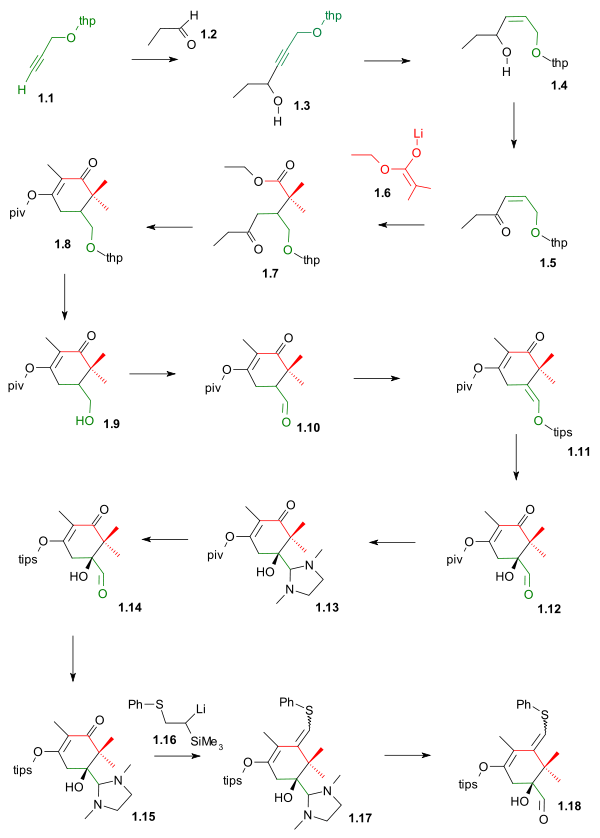 Kuwajime Taxol scheme 1 |
| Scheme 1 |
|---|
Synthesis B ring
The bottom part of ring B was constructed by nucleophilic addition to the aldehyde of 2.1 (scheme 2) with dibenzyl acetal of 2-bromobenzaldehyde 2.2 as its aryllithium. This step is much in common with the B ring synthesis in the Nicolaou Taxol total synthesis except that the aldehyde group is located at ring A and not ring B. The diol in 2.3 was protected as the boronic ester 2.4 preparing the molecule for upper part ring closure with tin tetrachloride to tricycle 2.5 in a Grob fragmentation-like reaction.
After deprotection (pinacol) to diol 2.6, DIBAL reduction to triol 2.7 and TBS reprotection (TBSOtf, lutidine) to alcohol 2.8 it was possible to remove the phenylsulfide group in with a tributyltin hydride and AIBN(see Barton-McCombie deoxygenation) to alcohol 2.9. Palladium on carbon hydrogenation removed the benzyl protecting group allowing the Swern oxidation of 2.10 to ketone 2.11
 Kuwajime Taxol scheme 2 |
| Scheme 2 |
|---|
Synthesis C ring
Completion of the C ring required complete reduction of the arene, placement of para oxygen atoms and importantly introduction of the C19 methyl group. The first assault on the aromatic ring in 3.1 (scheme 3) was launched with Birch reduction (potassium, ammonia, tetrahydrofuran, -78 °C, then ethanol) to diene 3.2. Deprotection (TBAF) to diol 3.3, reprotection as the benzaldehyde acetal 3.4 and reduction (sodium borohydride) to alcohol 3.5 allowed the oxidation of the diene to the 1,4-butenediol 3.6. In this photochemical [4+2]cycloaddition, singlet oxygen was generated from oxygen and rose bengal and the intermediate peroxide was reduced with thiourea. The next order of business was introduction of the C19 fragment: the new diol group was protected as the PMP acetal 3.7 (PMP stands for p-methoxyphenyl) allowing the oxidation of the C4 alcohol to ketone 3.8 with the Dess-Martin periodinane. Diethylaluminum cyanide reacted in a conjugate addition to the enone group to nitrile 3.9. The enol was protected as the TBS ether 3.10 allowing for the reduction of the nitrile group first to the aldehyde with DIBAL and then on to the alcohol 3.11 with Lithium aluminium hydride. The alcohol group was replaced by bromine in an Appel reaction which caused an elimination reaction (loss of HBr) to cyclopropane 3.12. Treatment with hydrochloric acid formed ketone 3.13, reaction with Samarium(II) iodide gave ring-opening finally putting the C19 methyl group in place in 3.14 and deprotection (TBAF) and enol-ketone conversion gave hydroxyketone 3.15
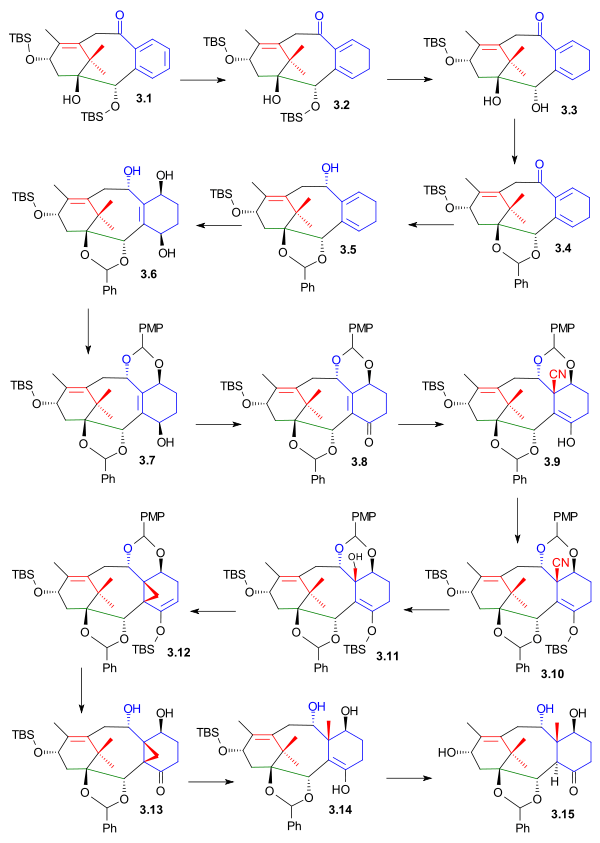 Kuwajime Taxol scheme 3 |
| Scheme 3 |
|---|
Synthesis D ring
By protecting the diol group in triol 4.1 (scheme 4) as the phenyl boronic ester 4.2, the remaining alcohol group could be protected as the TBS ether 4.3. After deprotecting the diol group (hydrogen peroxide, sodium bicarbonate) again in 4.4 it was possible to oxidize the C19 alcohol to the ketone 4.5 with Dess-Martin periodinane. In a new round of protections the C7 alcohol was converted to the 2-methoxy-2-propyl (MOP) ether 4.6 with 2-propenylmethylether and PPTS and the C7 ketone was converted to its enolate 4.7 by reaction with KHMDS and N,N-bis(trifluoromethylsulfonyl)aniline. These preambles facilitated the introduction of the final missing C20 fragment as the Grignard reagent trimethylsilylmethylmagnesium bromide which coupled with the triflate in a tetrakis(triphenylphosphine)palladium(0) catalysed reaction to the silane 4.8. The trimethylsilyl group eliminated on addition of NCS to organochloride 4.9. Prior to ring-closing the D ring there was some unfinished business in ring C. A C10 alcohol was introduced by MoOPH oxidation to 4.10 but with the wrong stereochemistry. After acetylation to 4.11 and inversion of configuration with added base DBN this problem was remedied in compound 4.12. Next dihydroxylation with Osmium(VIII) oxide formed the diol 4.13 with the primary alcohol on addition of base DBU displacing the chlorine atom in a nucleophilic aliphatic substitution to oxetane 4.14.
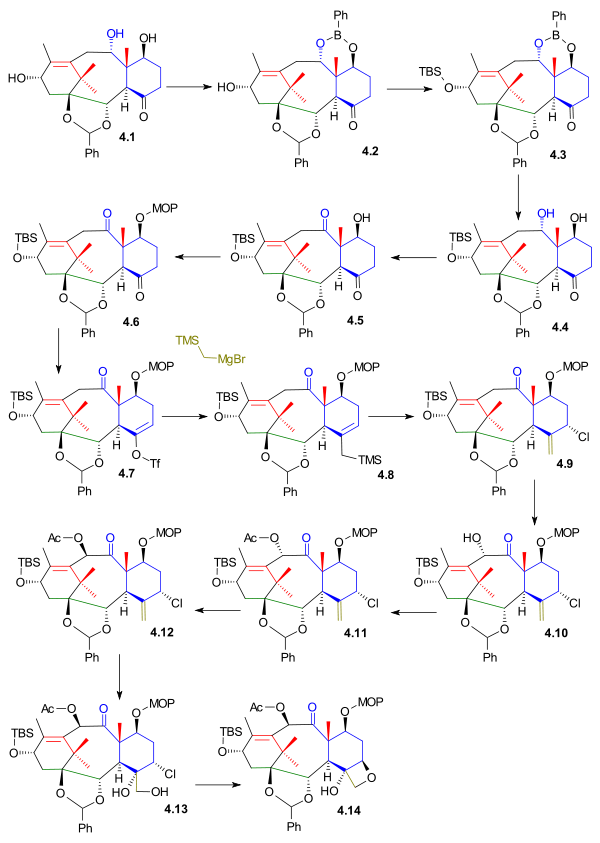 Kuwajime Taxol scheme 4 |
| Scheme 4 |
|---|
Tail addition
The C1, C2 and C4 functional groups were put in place next and starting from oxetane 5.1 (scheme 5) the MOM protecting group is removed in 5.2 (PPTS) and replaced by a TES group TESCl) in 5.3. The acetal group was removed in 5.4 (hydrogenation PdOH2, H2) and replaced by a carbonate ester group in 5.5 (triphosgene, pyridine). The tertiary alcohol group was acetylated in 5.6 and in the final step the carbonate group was opened by reaction with phenyllithium to the hydroxyester 5.7.
Prior to tail addition the TES protective group was removed in 5.8 (hydrogen fluoride pyridine) and replaced by a TROC (trichloroethyl carbonate, TROCCl ) group in 5.9. The C13 alcohol protective group was removed in 5.10 (TASF) enabling the tail addition of Ojima lactam 5.11 (this step is common with all total synthetic efforts to date) to 5.12 with Lithium bis(trimethylsilyl)amide. The synthesis was completed with TROC removal (zinc, acetic acid) to taxol 5.13.
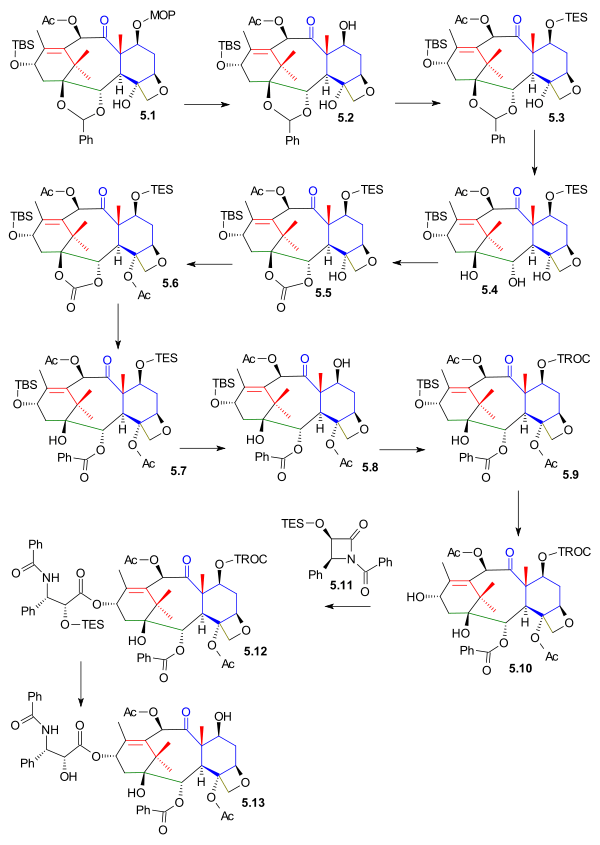 Kuwajime Taxol scheme 5 |
| Scheme 5 |
|---|
See also
External links
References
- Koichiro Morihira, Ryoma Hara; Soc, Isao Kuwajima; Kawahara, Shigeru; Nishimori, Toshiyuki; Nakamura, Nobuhito; Kusama, Hiroyuki; Kuwajima, Isao (1998). "Enantioselective Total Synthesis of Taxol". J. Am. Chem. Soc. 120 (49): 12980–12981. doi:10.1021/ja9824932.
- Hiroyuki Kusama; Ryoma Hara; Shigeru Kawahara; Toshiyuki Nishimori; Hajime Kashima; Nobuhito Nakamura; Koichiro Morihira; Isao Kuwajima (2000). "Enantioselective Total Synthesis of (−)-Taxol". J. Am. Chem. Soc. 122 (16): 3811–3820. doi:10.1021/ja9939439.