Wender Taxol total synthesis
The Wender Taxol total synthesis in organic chemistry describes a Taxol total synthesis (one of six to date) by the group of Paul Wender at Stanford University published in 1997.[1][2] This synthesis has much in common with the Holton Taxol total synthesis in that it is a linear synthesis starting from a naturally occurring compound with ring construction in the order A,B,C,D. The Wender effort is shorter by approximately 10 steps.
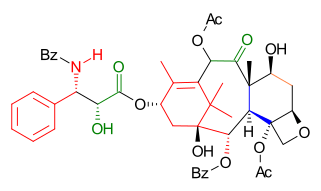
Raw materials for the preparation of Taxol by this route include verbenone, prenyl bromine, allyl bromide, propiolic acid, Gilman reagent, and Eschenmoser's salt.
AB Ring synthesis
The taxol synthesis started from the terpene verbenone 1 in Scheme 1, which is the oxidation product of naturally occurring α-pinene and forming ring A. Construction of ring B started with abstraction of the pendant methyl group proton by potassium tert-butoxide (conjugated anion is formed) followed by nucleophilic displacement of the bromine atom in prenyl bromide 2 to form diene 3. Ozonolysis of the prenyl group (more electron-rich than the internal double bond) formed aldehyde 4, which, after isomerization or photorearrangement to the chrysanthenone 5, was reacted with the lithium salt (via LDA) of the ethyl ester of propiolic acid 6 in a nucleophilic addition to the alcohol 7. This compound was not isolated but trapped in situ with trimethylsilyl chloride to the silyl ether 9. In the next step, Gilman reagent 8 is a methylating reagent in nucleophilic conjugate addition through the alkyne group to the ketone group, which formed the alcohol 10. The silyl ether protective group was removed by reaction with acetic acid to alcohol 11, which was then oxidized to the ketone 12 with RuCl2(PPh3)3 and NMO as the sacrificial catalyst. The acyloin group in 13 was introduced by KHMDS and Davis’ oxaziridine (see Holton Taxol total synthesis for another use of this system) and its hydroxyl group together with the ester group were reduced by lithium aluminium hydride to tetrol 14. Finally, the primary alcohol group was protected as a tert-butyldimethylsilyl ether by the corresponding silylchloride and imidazole in triol 15.
 Wender Taxol Scheme 1 |
| Scheme 1 |
|---|
In the second part (Scheme 2) the procedures are still confined to rings A and B. More protective groups were added to triol 15 as reaction with PPTS and 2-methoxypropene gives the acetonide 16. At this point the double bond in ring A was epoxidized with m-CPBA and sodium carbonate to epoxide 17 and a Grob fragmentation (also present in the Holton effort) initiated by DABCO opened up the AB ring system in alcohol 18, which was not isolated but protected as a TIPS silyl ether 19 with triisopropylsilyl triflate and 2,6-lutidine. The C1 position was next oxidized by the phosphite ester, P(OEt)3 and the strong base KOt-Bu, and oxygen to alcohol 20 (the stereochemistry controlled by bowl-shaped AB ring with hydroxylation from unhindered convex direction), the primary alcohol group was deprotected with ammonium chloride in methanol to diol 21 and two reductions first with NaBH4 to triol 22 and then hydrogen gas and Crabtree's catalyst give triol 23. These positions were protected by trimethylsilyl chloride and pyridine to 24 and then triphosgene to 25 in order to facilitate the oxidation of the primary alcohol group to the aldehyde 26 by PCC.
 Wender Taxol Scheme 2 |
| Scheme 2 |
|---|
C Ring synthesis
The next part constructed the C ring starting from aldehyde 26, which was extended by one carbon atom to homologue 27 in a Wittig reaction with Methoxymethylenetriphenylphosphine (Scheme 3). The acetonide group was removed by dilute hydrochloric acid and sodium iodide in dioxane and one hydroxyl group in the resulting diol 28 was protected as the triethylsilyl ether (TES) 29 with the corresponding silyl chloride and pyridine enabling oxidation of the remaining hydroxyl group to the ketone 30 with the Dess-Martin periodinane. Reaction with Eschenmoser's salt placed a methylene group (C20 in the Taxol framework) in the alpha position of the aldehyde to 31 and the next reaction introduced (the still lacking) C6 and C7 as the Grignard reagent of allyl bromide in a nucleophilic addition aided by zinc(II) chloride, which blocked the Grignard from attack on carbonate group, to alcohol 32. The newly formed alcohol was protected as the BOM ether 33 with BOMCl and N,N-diisopropylethylamine. After removal of the TES protecting group with ammonium fluoride, the carbonate group in 34 was converted to a hydroxybenzoate group by action of phenyllithium and the secondary alcohol to the acetate 35 by in situ reaction with acetic anhydride and DMAP. In the next step the acyloin group had its positions swapped by reaction with triazabicyclodecene (other amine bases fail) forming 36 and in the final steps ring closure of ring C was accomplished by ozonolysis at the allyl group to 37 and Aldol reaction with 4-pyrrolidinopyridine to 38.
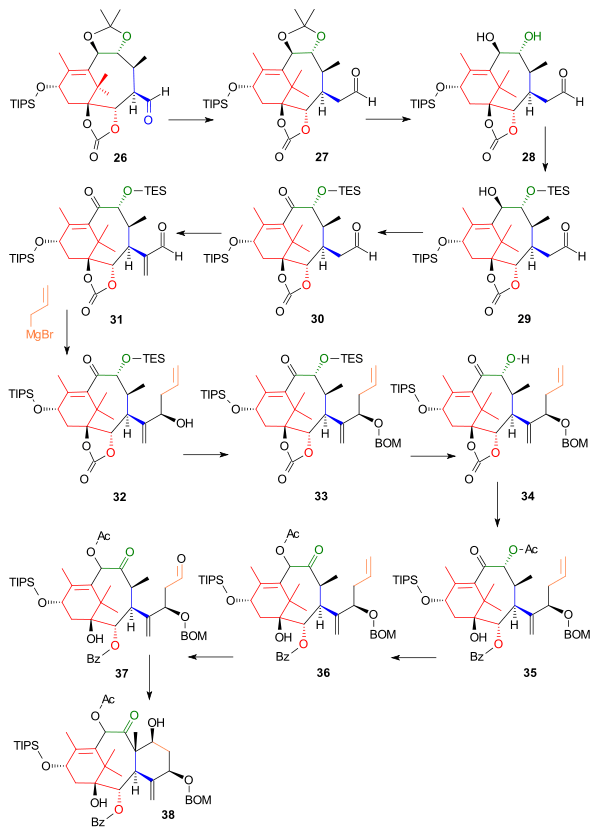 Wender Taxol Scheme 3 |
| Scheme 3 |
|---|
D Ring synthesis
The final part dealt with the construction of oxetane ring D starting with protection of the alcohol group in 38 (Scheme 4). as a TROC alcohol 39 with 2,2,2-trichloroethyl chloroformate and pyridine. The OBOM group was replaced by a bromine group in three steps: deprotection to 40 with hydrochloric acid and sodium iodide, mesylation to 41 with mesyl chloride, DMAP and pyridine and nucleophilic substitution with inversion of configuration with lithium bromide to bromide 42. Because the oxidation of the alkene group to the diol 43 with osmium tetroxide was accompanied by the undesired migration of the benzoate group, this step was taken to completion with imidazole as 44. Two additional countermeasures were required: reprotection of the diol as the carbonate ester 45 with triphosgene and removal of the benzoate group (KCN) to alcohol 46 in preparation of the actual ring closure to the oxetane 47 with N,N-diisopropylethylamine. In the final steps the tertiary alcohol was acylated in 48, the TIPS group removed in 49 and the benzoate group re-introduced in 50.
Tail addition of the Ojima lactam 51 was not disclosed in detail but finally taxol 52 was formed in several steps similar to the other efforts.
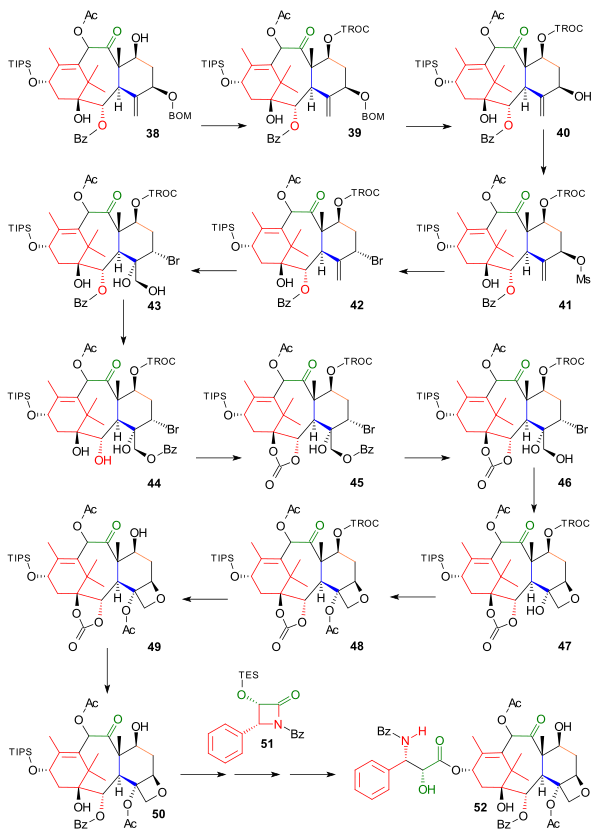 Wender Taxol Scheme 4 |
| Scheme 4 |
|---|
External links
- Wender Taxol Synthesis @ SynArchive.com
- The Wender Taxol Mug: Link
See also
References
- The Pinene Path to Taxanes. 5. Stereocontrolled Synthesis of a Versatile Taxane Precursor Paul A. Wender et al.J. Am. Chem. Soc.; 1997; 119(11) pp 2755 - 2756; (Communication) doi:10.1021/ja9635387
- The Pinene Path to Taxanes. 6. A Concise Stereocontrolled Synthesis of Taxol Wender, P. A. et al. J. Am. Chem. Soc.; (Communication); 1997; 119(11); 2757-2758. doi:10.1021/ja963539z