Holton Taxol total synthesis
The Holton Taxol total synthesis, published by Robert A. Holton and his group at Florida State University in 1994 was the first total synthesis of Taxol (generic name: paclitaxel).[1][2]
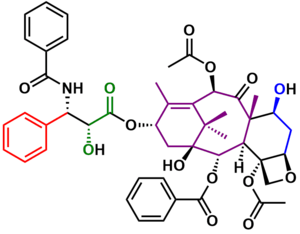
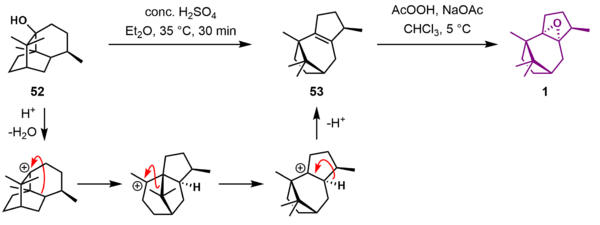
The Holton Taxol total synthesis is a good example of a linear synthesis starting from commercially available natural compound patchoulene oxide.[3] This epoxide can be obtained in two steps from the terpene patchoulol and also from borneol.[4][5] The reaction sequence is also enantioselective, synthesizing (+)-Taxol from (−)-patchoulene oxide or (−)-Taxol from (−)-borneol with a reported specific rotation of +- 47° (c=0.19 / MeOH). The Holton sequence to Taxol is relatively short compared to that of the other groups (46 linear steps from patchoulene oxide). One of the reasons is that patchoulene oxide already contains 15 of the 20 carbon atoms required for the Taxol ABCD ring framework.
Other raw materials required for this synthesis include 4-pentenal, m-chloroperoxybenzoic acid, methyl magnesium bromide and phosgene. Two key chemical transformations in this sequence are a Chan rearrangement and a sulfonyloxaziridine enolate oxidation.
Retrosynthesis
It was envisaged that Taxol (51) could be accessed through tail addition of the Ojima lactam 48 to alcohol 47. Of the four rings of Taxol, the D ring was formed last, the result of a simple intramolecular SN2 reaction of hydroxytosylate 38, which could be synthesized from hydroxyketone 27. Formation of the six-membered C ring took place through a Dieckmann condensation of lactone 23, which could be obtained through a Chan rearrangement of carbonate ester 15. Substrate 15 could be derived from ketone 6, which, after several oxidations and rearrangements, could be furnished from commercially available patchoulene oxide 1.
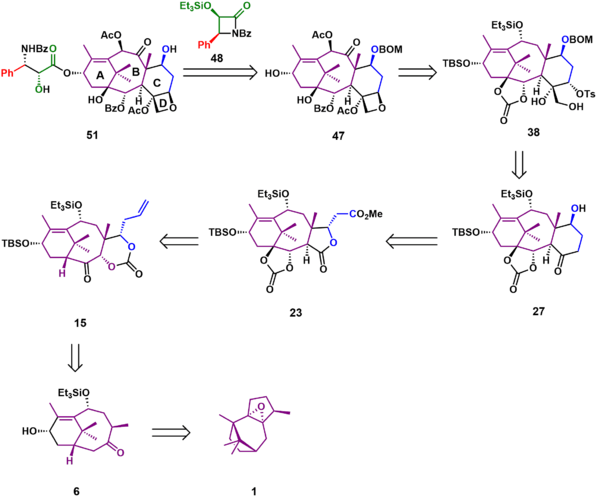
AB Ring synthesis
As shown in Scheme 1, the first steps in the synthesis created the bicyclo[5.3.1]undecane AB ring system of Taxol. Reaction of epoxide 1 with tert-butyllithium removed the acidic α-epoxide proton, leading to an elimination reaction and simultaneous ring-opening of the epoxide to give allylic alcohol 2. The allylic alcohol was epoxidized to epoxyalcohol 3 using tert-butyl hydroperoxide and titanium(IV)tetraisopropoxide. In the subsequent reaction, the Lewis acid boron trifluoride catalyzed the ring opening of the epoxide followed by skeletal rearrangement and an elimination reaction to give unsaturated diol 4. The newly created hydroxyl group was protected as the triethylsilyl ether (5). A tandem epoxidation with meta-chloroperbenzoic acid and Lewis acid-catalyzed Grob fragmentation gave ketone 6, which was then protected as the tert-butyldimethylsilyl ether 7 in 94% yield over three steps.
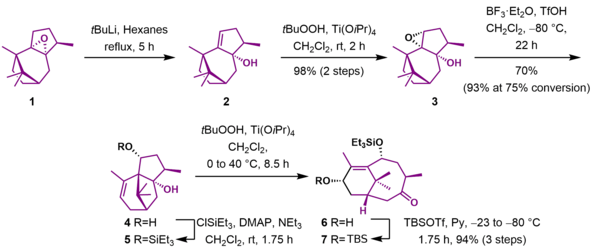
C Ring preparation
As shown in Scheme 2, the next phase involved addition of the carbon atoms required for the formation of the C ring. Ketone 7 was treated with magnesium bromide diisopropylamide and underwent an aldol reaction with 4-pentanal (8) to give β-hydroxyketone 9. The hydroxyl group was protected as the asymmetric carbonate ester (10). Oxidation of the enolate of ketone 10 with (-)-camphorsulfonyl oxaziridine (11) gave α-hydroxyketone 12. Reduction of the ketone group with 20 equivalents of sodium bis(2-methoxyethoxy)aluminumhydride (Red-Al) gave triol 13, which was immediately converted to carbonate 14 by treatment with phosgene. Swern oxidation of alcohol 14 gave ketone 15. The next step set the final carbon-carbon bond between the B and C rings. This was achieved through a Chan rearrangement of 15 using lithium tetramethylpiperidide to give α-hydroxylactone 16 in 90% yield. The hydroxyl group was reductively removed using samarium(II) iodide to give an enol, and chromatography of this enol on silica gel gave the separable diastereomers cis 17c (77%) and trans 17t (15%), which could be recycled to 17c through treatment with potassium tert-butoxide. Treatment of pure 17c with lithium tetramethylpiperidide and (±)-camphorsulfonyl oxaziridine gave separable α-hydroxyketones 18c (88%) and 18t (8%) in addition to some recovered starting material (3%). Reduction of pure ketone 18c using Red-Al followed by basic work-up resulted in epimerization to give the required trans-fused diol 19 in 88% yield.
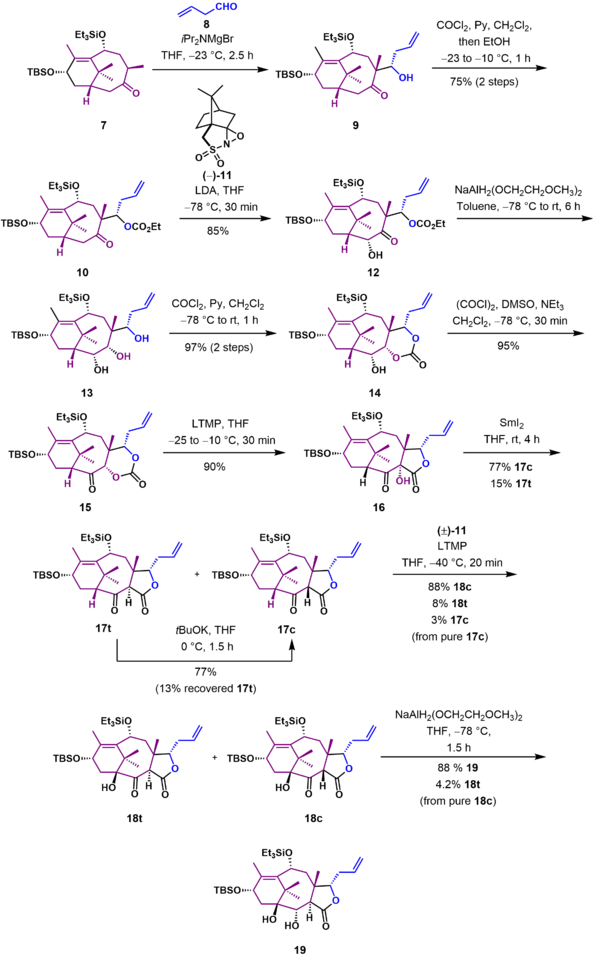
C Ring synthesis
As shown in Scheme 3, diol 19 was protected with phosgene as a carbonate ester (20). The terminal alkene group of 20 was next converted to a methyl ester using ozonolysis followed by oxidation with potassium permanganate and esterification with diazomethane. Ring expansion to give the cyclohexane C ring 24 was achieved using a Dieckman condensation of lactone 23 with lithium diisopropylamide as a base at -78 °C. Decarboxylation of 24 required protection of the hydroxyl group as the 2-methoxy-2-propyl (MOP) ether (25). With the protecting group in place, decarboxylation was effected with potassium thiophenolate in dimethylformamide to give protected hydroxy ketone 26. In the next two steps the MOP protecting group was removed under acidic conditions, and alcohol 27 was reprotected as the more robust benzyloxymethyl ether 28. The ketone was converted to the trimethylsilyl enol ether 29, which was subsequently oxidized in a Rubottom oxidation using m-chloroperbezoic acid to give the trimethylsilyl protected acyloin 30. At this stage the final missing carbon atom in the Taxol ring framework was introduced in a Grignard reaction of ketone 30 using a 10-fold excess of methylmagnesium bromide to give tertiary alcohol 31. Treatment of this tertiary alcohol with the Burgess reagent (32) gave exocyclic alkene 33.
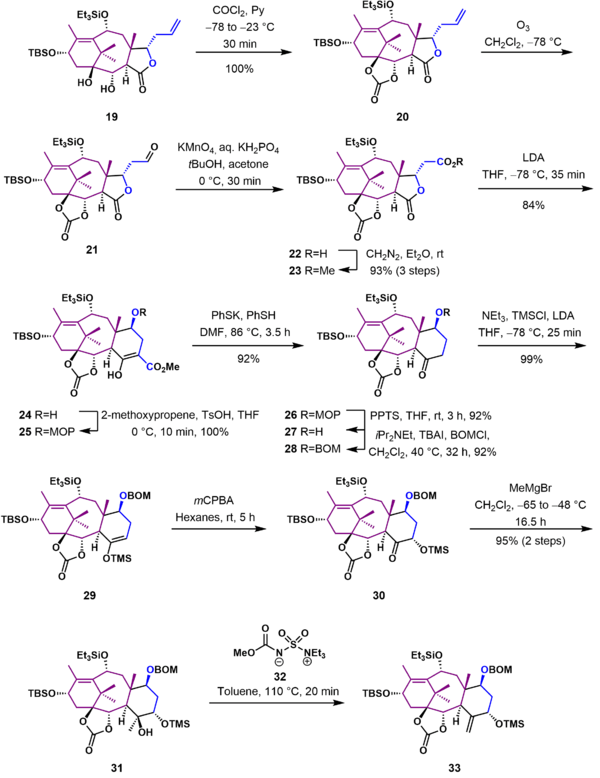
D Ring synthesis and AB ring elaboration
In this section of the Holton Taxol synthesis (Scheme 4), the oxetane D ring was completed and ring B was functionalized with the correct substituents. Allylic alcohol 34, obtained from deprotection of silyl enol ether 33 with hydrofluoric acid, was oxidized with osmium tetroxide in pyridine to give triol 35. After protection of the primary hydroxyl group, the secondary hydroxyl group in 36 was converted to a good leaving group using p-toluenesulfonyl chloride. Subsequent deprotection of the trimethylsilyl ether 37 gave tosylate 38, which underwent cyclization to give oxetane 39 by nucleophilic displacement of the tosylate that occurred with inversion of configuration. The remaining unprotected tertiary alcohol was acylated, and the triethylsilyl group was removed to give allylic alcohol 41. The carbonate ester was cleaved by reaction with phenyllithium in tetrahydrofuran at -78 °C to give alcohol 42. The unprotected secondary alcohol was oxidized to ketone 43 using tetrapropylammonium perruthenate (TPAP) and N-methylmorpholine N-oxide (NMO). This ketone was deprotonated with potassium tert-butoxide in tetrahydrofuran at low temperature and further oxidized by reaction with benzeneseleninic anhydride to give α-hydroxyketone 44. Further treatment of 44 with potassium tert-butoxide furnished α-hydroxyketone 45 through a Lobry-de Bruyn-van Ekenstein Rearrangement. Substrate 45 was subsequently acylated to give α-acetoxyketone 46.
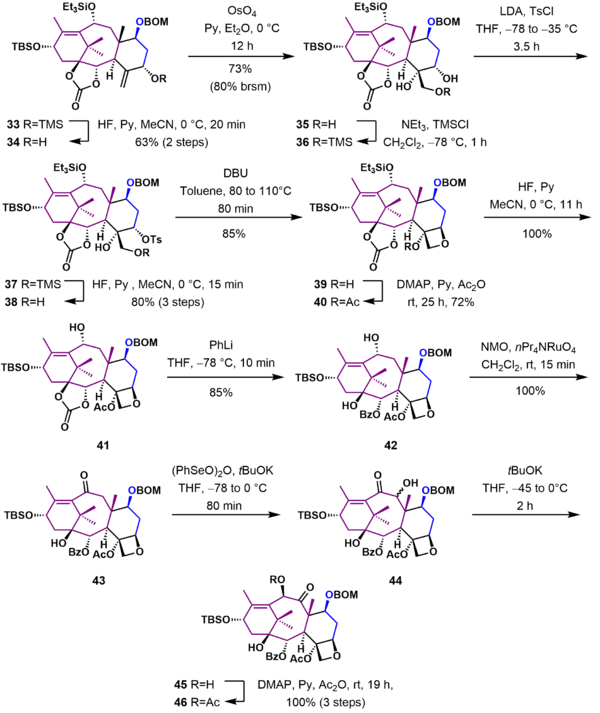
Tail addition
In the final stages of the synthesis (Scheme 5), the hydroxyl group in 46 was deprotected to give alcohol 47. Reaction of the lithium alkoxide of 47 with the Ojima lactam 48 adds the tail in 49. Deprotection of the triethylsilyl ether with hydrofluoric acid and removal of the BOM group under reductive conditions gave (−)-Taxol 51 in 46 steps.
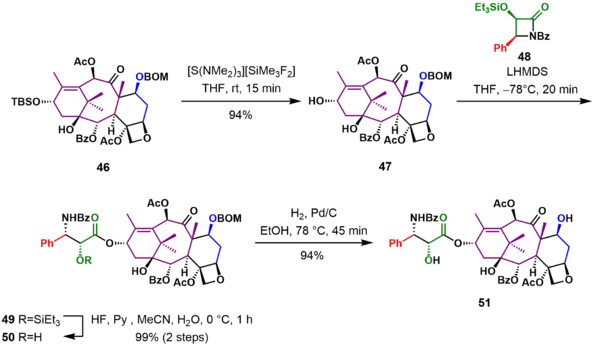
Precursor synthesis
Patchoulene oxide (1) could be accessed from terpene patchoulol (52) through a series of acid-catalyzed carbocation rearrangements proceeded by an elimination following Zaitzev's rule to give pathoulene (53). The driving force for the rearrangement is relief of ring strain. Epoxidation of 53 with peracetic acid gave patchoulene oxide 1.
Protecting Groups
BOM (benzyloxymethyl)
Protection Reagents: Benzyloxymethyl chloride, N,N-diisopropylethanamine, tetrabutylammonium iodide, in refluxing dichloromethane, 32 h.
Deprotection Reagents: H2, Pd/C
Alcohol 27 (Scheme 3) was protected as the BOM ether, a more robust protecting group than MOP (see below).
Carbonate (asymmetric)
Protection Reagents: Phosgene, pyridine, ethanol in dichloromethane, -23 to -10 °C.
Deprotection Reagents: Sodium bis(2-methoxyethoxy)aluminumhydride (Red-Al)
The secondary alcohol in the 4-pentenal product of the aldol reaction, 9, was protected as an asymmetric carbonate ester. This group was removed in conjunction with the Red-Al reduction of ketone 12 (Scheme 2).
Carbonate (cyclic) [1]
Protection Reagent: Phosgene, pyridine, dichloromethane, -78 °C to room temperature, 1 h.
Deprotection Reagents: deprotected through Chan rearrangement (treatment with lithiumtetramethylpiperidide).
The cyclic carbonate ester was removed as a result of the Chan rearrangement in 15, which created a carbon-carbon bond that was part of the Taxol framework (Scheme 2).
Carbonate (cyclic) [2]
Protection Reagent: Phosgene, pyridine, -78 to -23 °C, 0.5 h
Deprotection Reagents: Phenyllithium in tetrahydrofuran at -78 °C.
Diol 19 (Scheme 3) was protected as a cyclic carbonate ester. This carbonate ester was cleaved by phenyllithium in tetrahydrofuran at -78 °C to give hydroxybenzoate 42 (Scheme 4).
MOP (2-methoxy-2-propyl)
Protection Reagents: p-Toluenesulfonic acid and 2-methoxypropene
Deprotection Reagents: Tetrabutylammonium fluoride (1 mol eq., THF, -1 °C, 6 h)
The hydroxyl group in hydroxyester 24 (Scheme 3) was protected as a MOP ether in order to decarboxylate the β-ketoester group.
TBS (tert-butyldimethylsilyl)
Protection Reagents: Butyllithium, tetrahydrofuran, tert-butyldimethylsilyl chloride
Deprotection Reagents: Tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF)
After Grob fragmentation (Scheme 1), the resultant alcohol 6 was protected as a TBS ether 7, which is kept in place until the final addition of the tail (Scheme 5).
TES (triethylsilyl) [1]
Protection Reagents: Triethylsilyl chloride, 4-(dimethylamino)pyridine, pyridine
Deprotection Reagents: Hydrogen fluoride/pyridine complex in acetonitrile
The secondary hydroxyl group in diol 4 (Scheme 1) was protected as a TES ether in order to prevent its participation in the Grob fragmentation. The TES was cleaved in 37 (Scheme 4) and returned to the alcohol.
TES (triethylsilyl) [2]
Protection Reagents: See Ojima lactam
Deprotection Reagents: Hydrogen fluoride, pyridine, acetonitrile, 0 °C, 1 h
The secondary alcohol of 48 (Scheme 5) needed to be protected until addition of the tail to the secondary hydroxyl group in ring A was complete.
TMS (trimethylsilyl) [1]
Protection Reagents: Lithium diisopropylamide, trimethylsilyl chloride
Deprotection Reagents: Hydrofluoric acid, pyridine, acetonitrile.
Ketone 25 (Scheme 3) was protected as the TMS enol ether and subsequently was oxidized with M-chloroperoxybenzoic acid. In the process the TMS group migrated to the 2-hydroxyl group.
TMS (trimethylsilyl) [2]
Protection Reagents: Trimethylsilyl chloride
Deprotection Reagents: Hydrofluoric acid, pyridine, acetonitrile
The primary hydroxyl group in triol 35 (Scheme 4) was protected as a TMS ether allowing activation of the secondary hydroxyl group as a tosylate leaving group.
See also
References
- Robert A. Holton; Carmen Somoza; Hyeong Baik Kim; Feng Liang; Ronald J. Biediger; P. Douglas Boatman; Mitsuru Shindo; Chase C. Smith; Soekchan Kim; Hossain Nadizadeh; Yukio Suzuki; Chunlin Tao; Phong Vu; Suhan Tang; Pingsheng Zhang; Krishna K. Murthi; Lisa N. Gentile; Jyanwei H. Liu (1994). "First total synthesis of taxol. 1. Functionalization of the B ring". J. Am. Chem. Soc. 116 (4): 1597–1598. doi:10.1021/ja00083a066.
- Robert A. Holton; Hyeong-Baik Kim; Carmen Somoza; Feng Liang; Ronald J. Biediger; P. Douglas Boatman; Mitsuru Shindo; Chase C. Smith; Soekchan Kim; Hossain Nadizadeh; Yukio Suzuki; Chunlin Tao; Phong Vu; Suhan Tang; Pingsheng Zhang; Krishna K. Murthi; Lisa N. Gentile; Jyanwei H. Liu (1994). "First Total Synthesis of Taxol. 2. Completion of the C and D Rings". J. Am. Chem. Soc. 116 (4): 1599–1600. doi:10.1021/ja00083a067.
- Robert A. Holton; R. R. Juo; Hyeong B. Kim; Andrew D. Williams; Shinya. Harusawa; Richard E. Lowenthal; Sadamu. Yogai (1988). "A synthesis of taxusin". J. Am. Chem. Soc. 110 (19): 6558–6560. doi:10.1021/ja00227a043.
- Buchi, G.; MacLeod, William D.; Padilla, J. (1964-10-01). "Terpenes. XIX.1 Synthesis of Patchouli Alcohol2". Journal of the American Chemical Society. 86 (20): 4438–4444. doi:10.1021/ja01074a041. ISSN 0002-7863.
- Büchi, G.; Erickson, R. E.; Wakabayashi, Nobel (1961-02-01). "Terpenes. XVI.1,2 Constitution of Patchouli Alcohol and Absolute Configuration of Cedrene". Journal of the American Chemical Society. 83 (4): 927–938. doi:10.1021/ja01465a042. ISSN 0002-7863.