Nicolaou Taxol total synthesis
The Nicolaou Taxol total synthesis, published by K. C. Nicolaou and his group in 1994 concerns the total synthesis of Taxol.[1] Taxol is an important drug in the treatment of cancer but also expensive because the compound is harvested from a scarce resource, namely the pacific yew.
This synthetic route to taxol is one of several; other groups have presented their own solutions, notably the group of Holton with a linear synthesis starting from borneol, the Samuel Danishefsky group starting from the Wieland-Miescher ketone and the Wender group from pinene.
The Nicolaou synthesis is an example of convergent synthesis because the molecule is assembled from three pre-assembled synthons. Two major parts are cyclohexene rings A and C that are connected by two short bridges creating an 8 membered ring in the middle (ring B). The third pre-assembled part is an amide tail. Ring D is an oxetane ring fused to ring C. Two key chemical transformations are the Shapiro reaction and the pinacol coupling reaction.[2] The overall synthesis was published in 1995 in a series of four papers.[3][4][5][6]
Retrosynthesis
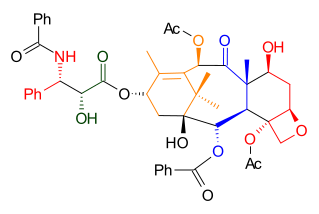
As illustrated in Retrosynthetic Scheme I, Taxol was derived from diol 7.2 by an ester bond formation, according to the Ojima-Holton method. This diol comes from carbonate 6.3 by the addition of phenyllithium. The oxetane ring in compound 6.3 was obtained via an SN2 reaction involving a mesylate derived from acetal 4.9. Ring B was closed via a McMurry reaction involving dialdehyde 4.8 which ultimately was derived from aldehyde 4.2 and hydrazone 3.6 using a Shapiro coupling reaction.
 Scheme 1 Nicolaou Taxol Retrosynthesis |
| Retrosynthesis Scheme 1 |
|---|
Retrosynthetic Scheme II indicates that both the aldehyde and the hydrazone used in the Shapiro coupling reaction were synthesized using Diels-Alder reactions.
 Scheme 2 Nicolaou Retrosynthesis |
| Retrosynthesis Scheme 2 |
|---|
C Ring synthesis
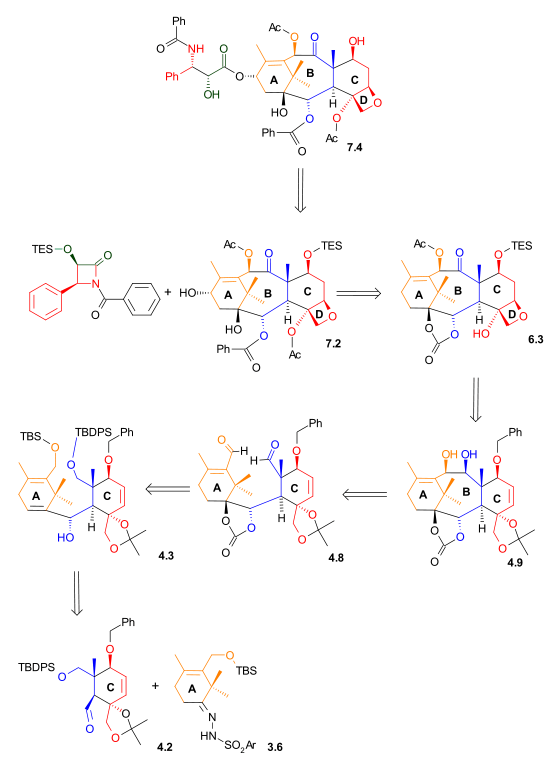
As shown in Scheme 1, the ring synthesis of ring C began with a Diels-Alder reaction between diene 1.3 and dienophile 1.1 in the presence of phenylboronic acid (1.2), which, after addition of 2,2-dimethyl-1,3-propanediol, gave five-membered lactone 1.8 in 62% yield. Boron served as a molecular tether and aligned both diene and dienophile for this endo Diels-Alder cycloaddition. After protection of the hydroxyl groups as tert-butyldimethylsilyl ethers, reduction of the ester with lithium aluminium hydride and selective deprotection of the secondary hydroxyl group gave lactone diol 1.11. The unusual lactone hydrates 1.9 and 1.10 were isolated as synthetic intermediates in this process.
 Ring C synthesis Scheme 1 |
| Scheme 1 |
|---|
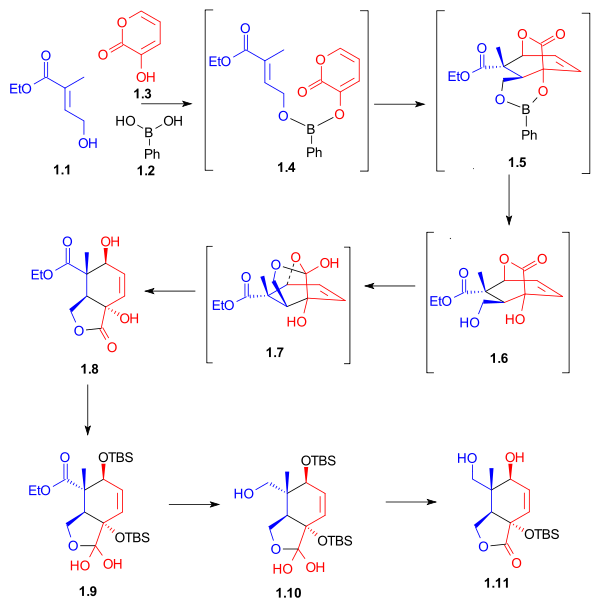
Lactone diol 2.1, after selective protection, was reduced with lithium aluminium hydride to give triol 2.4. This triol, after convas the acetonide, was selectively oxidized to the aldehyde using tetrapropylammonium perruthenate (TPAP) and N-methylmorpholine N-oxide. Aldehyde 2.6 served as a starting point for the construction of ring B (Scheme 4, compound 4.2).
 Ring C synthesis Scheme 2 |
| Scheme 2 |
|---|
A Ring synthesis
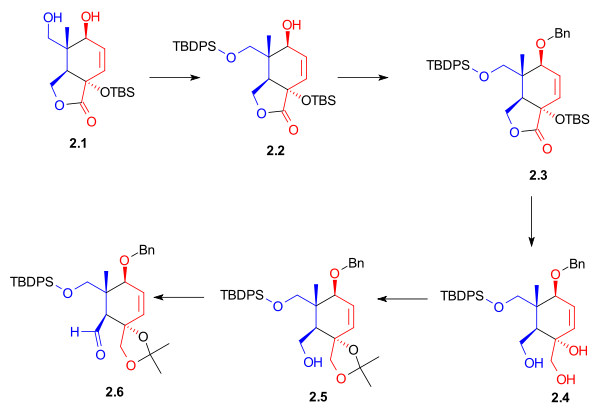
The A ring synthesis (Scheme 3) started with a Diels-Alder reaction of diene 3.1 with the commercially available dienophile 2-chloroacrylonitrile 3.2 to give cyclohexene 3.3 with complete regioselectivity. Hydrolysis of the cyanochloro group and simultaneous cleavage of the acetate group led to hydroxyketone 3.4. The hydroxyl group was protected as a tert-butyldimethylsilyl ether (3.5). In preparation for a Shapiro reaction, this ketone was converted to hydrazone 3.6.
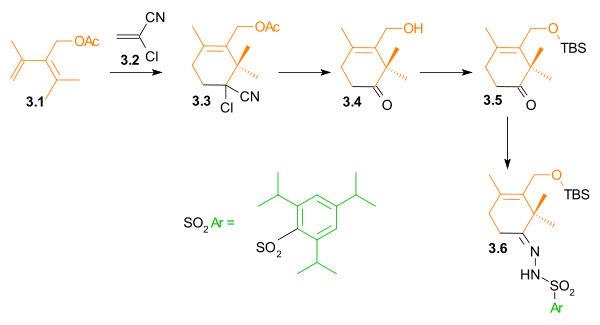 Ring A synthesis Scheme 3 |
| Scheme 3 |
|---|
B Ring synthesis
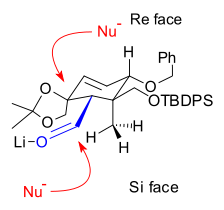
The coupling of ring A and ring C created the 8 membered B ring. One connection was made via a nucleophilic addition of a vinyllithium compound to an aldehyde and the other connection through a pinacol coupling reaction of two aldehydes (Scheme 4).
A Shapiro reaction of the vinyllithium compound derived from hydrazone 4.1 with aldehyde 4.2 makes the first connection that will become the B ring. The control of stereochemistry in 4.3 is thought to be derived from the relative hindrance of the Si face in the orientation shown on the right, due to the proximity of the axial methyl group. Epoxidation with vanadyl(acetylacetate) converted alkene 4.3 to epoxide 4.4, which, upon reduction with lithium aluminium hydride, gave diol 4.5. This diol was then protected as carbonate ester 4.6. The carbonate group also served to create rigidity in the ring structure for the imminent pinacol coupling reaction. The two silyl ether groups were removed, and diol 4.7 was then oxidized to give dialdehyde 4.8 using N-methylmorpholine N-oxide in the presence of a catalytic amount of tetrapropylammonium perruthenate. In the final step of the formation of Ring B, a pinacol coupling using conditions developed by McMurry (titanium(III) chloride and a zinc/copper alloy) gave diol 4.9.
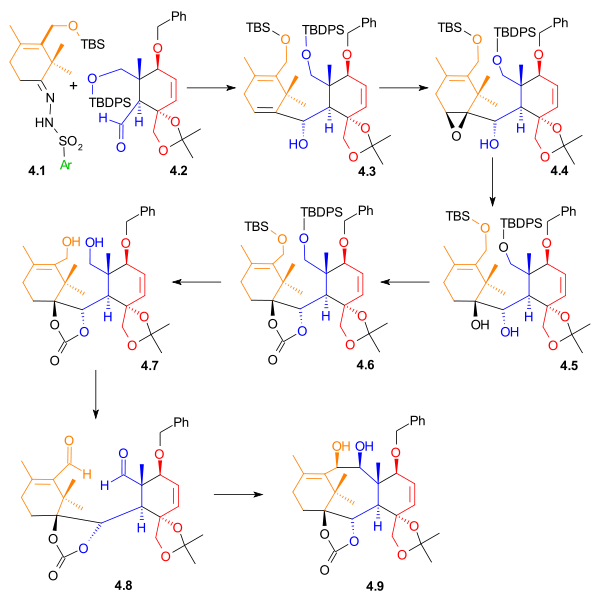 Ring B synthesis Scheme 4 |
| Scheme 4 |
|---|
Resolution
At this point in the synthesis of Taxol, the material was a racemic mixture. To obtain the desired enantiomer, allylic alcohol 4.9 was acylated with (1S)-(−)-camphanic chloride and dimethylaminopyridine, giving two diastereomers. These were then separated using standard column chromatography. The desired enantiomer was then isolated when one of the separated disatereomers was treated with potassium bicarbonate in methanol.
 resolution |
| Enantiomeric resolution of 4.9. |
D Ring synthesis
The desired enantiomer from resolution, allylic alcohol 5.1 (Scheme 5) was acetylated with acetic anhydride and 4-(dimethylamino)pyridine in methylene chloride to yield monoacetate 5.2. It is noteworthy that this reaction was exclusive for the allylic alcohol, and the adjacent hydroxyl group was not acetylated. Alcohol 5.2 was oxidized with tetrapropylammonium perruthenate and N-methylmorpholine N-oxide to give ketone 5.3. Alkene 5.3 underwent hydroboration in tetrahydrofuran. Oxidation with basic hydrogen peroxide and sodium bicarbonate gave alcohol 5.4 in 35% yield, with 15% yield of a regioisomer. The acetonide was removed, giving triol 5.5. This alcohol was monoacetylated, to give acetate 5.6. The benzyl group was removed and replaced with a triethylsilyl group. Diol 5.7 was selectively activated using methanesulfonyl chloride and 4-(dimethylamino)pyridine to give mesylate 5.8, in 78% yield.
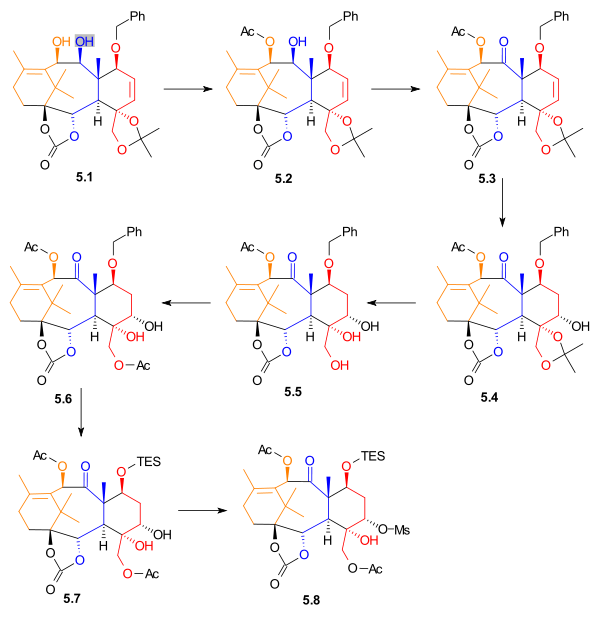 Ring D1 synthesis Scheme 5 |
| Scheme 5 |
|---|
The acetyl group in 6.1 (Scheme 6) was removed to give primary alcohol 6.2. The Taxol ring (D) was added by an intramolecular nucleophilic substitution involving this hydroxyl group to give oxetane 6.3. After acetylation, phenyllithium was used to open the carbonate ester ring to give alcohol 6.5. Allylic oxidation with pyridinium chlorochromate, sodium acetate, and celite gave ketone 6.6, which was subsequently reduced using sodium borohydride to give secondary alcohol 6.7. This was the last compound before the addition of the amide tail.
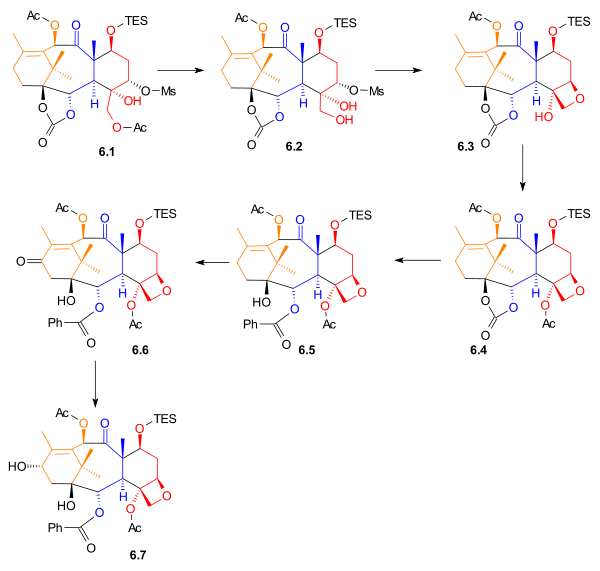 Ring D2 synthesis Scheme 6 |
| Scheme 6 |
|---|
Tail addition
As shown in Scheme 7, Ojima lactam 7.1 reacted with alcohol 7.2 with sodium bis(trimethylsilyl)amide as a base. This alcohol is the triethylsilyl ether of the naturally occurring compound baccatin III. The related compound, 10-deacetylbaccatin III, is found in Taxus baccata, also known as the European Yew, in concentrations of 1 gram per kilogram leaves. Removal of the triethylsilyl protecting group gave Taxol.
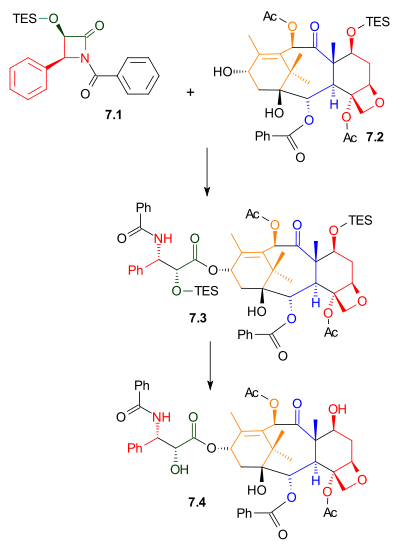 Tail Addition Scheme 7 |
| Scheme 7 |
|---|
Precursor synthesis
Synthesis of the Diels-Alder dienophile for Ring C
The ethyl ester of propionic acid (1) was brominated and then converted to the Wittig reagent using triphenylphosphine. Aldehyde 6 was obtained from allyl alcohol (4) by protection as the tert-butyldiphenylsilyl ether (5) followed by ozonolysis. Wittig reagent 3 and aldehyde 6 reacted in a Wittig reaction to give unsaturated ester 7, which was deprotected to give dienophile 8 (Scheme 1, compound 1).
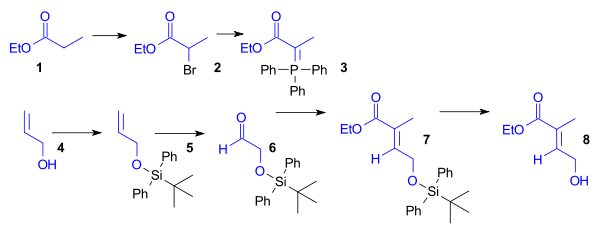
Synthesis of the Diels-Alder diene for Ring A
Aldol condensation of acetone and ethyl acetoacetate gave β-keto-ester 3. A Grignard reaction involving methylmagnesium bromide provided alcohol 4, which was subjected to acid catalyzed elimination to give diene 5. Reduction and acylation gave diene 7 (Scheme 3, compound 1).
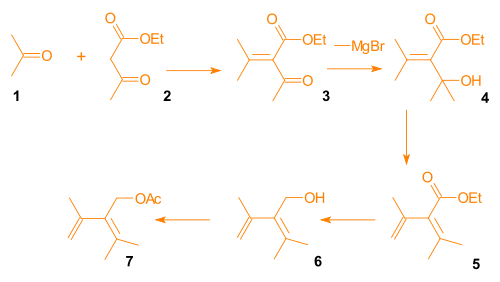
Protecting groups
Ac (acetyl)
Protection: Acetic anhydride, pyridine, 4-(dimethylamino)pyridine, and dichloromethane
Deprotection: Potassium carbonate in methanol and water solvent
Protection prevented mesylation of the primary oxygen in 5.8.
Acetonide
Protection: 2,2-dimethoxypropane and camphorsulfonic acid, and dichloromethane
Deprotection: Hydrochloric acid, methanol, water, and ethyl ether
Protection of the vicinal diol 2.4 allowed the remaining hydroxyl group in alcohol 2.5 to be selectively oxidized to give aldehyde 2.6. The acetonide was removed much later in the synthesis in preparation for the closure of ring D.
Bn (benzyl)
Protection: Potassium hydride, tetra-n-butylammonium iodide, and benzyl bromide.
Deprotection: Hydrogen, Pd(OH)2/C
Secondary alcohol 2.2 was protected as the benzyl ether so that the reduction of lactone 2.3 could occur. Protection was removed much later in the synthesis to form alcohol 5.7, which was reprotected as the triethylsilyl ether.
Carbonate Ester
Protection: Potassium hydride, phosgene
Deprotection: Phenyllithium opens the carbonate ester ring to give alcohol 6.5.
Protection adds rigidity in ring structure for pinacol coupling reaction forming diol 4.9, and also prevents unwanted oxidation in the formation of dialdehyde 4.8.
Protection: Tert-butyldiphenylsilyl chloride, imidazole, and dimethylformamide.
Deprotection: Tetra-n-butylammonium fluoride
Primary alcohol 2.1 was protected in preparation for lactone reduction in 2.3. The protecting group was removed to give diol 4.7 in preparation for the pinacol coupling reaction.
TBS (tert-butyldimethylsilyl) [1]
Protection: Tert-butyldimethylsilyl triflate, lutidine, 4-(dimethylamino)pyridine, and dichloromethane.
Deprotection: Camphorsulfonic acid, dichloromethane, and methanol.
The secondary hydroxyl group in 1.8 was briefly protected during the protection of a tertiary hydroxyl group in the same compound.
TBS (tert-butyldimethylsilyl) [2]
Protection: Tert-butyldimethylsilyl triflate, lutidine, 4-(dimethylamino)pyridine, and dichloromethane.
Deprotection: Camphorsulfonic acid
Protection of the tertiary hydroxyl group in 1.8 was necessary to allow selective protection of other hydroxyl groups on the C ring.
TBS (tert-butyldimethylsilyl) [3]
Protection: Dichloromethane, imidazole, and tert-butyldimethylsilyl chloride.
Deprotection: Tetra-n-butylammonium fluoride
Protection of hydroxyl group in 3.4 allowed the ketone to undergo a Shapiro reaction to form the viyllithium compound 3.7.
TES (triethylsilyl) [1]
Protection: Triethylsilyl chloride and pyridine.
Deprotection: Hydrolysis using hydrofluoric acid, pyridine and tetrahydrofuran.
Protection of the secondary hydroxyl group in 5.7 was necessary for the final addition of the tail to alcohol 7.2.
TES (triethylsilyl) [2]
Protection: See Ojima lactam.
Deprotection: Hydrolysis using hydrofluoric acid and pyridine
Protected secondary alcohol in Ojima lactam 7.1 during reaction with alcohol 7.2 in the tail addition.
See also
External links
References
- Classics in Total Synthesis: Targets, Strategies, Methods K. C. Nicolaou, E. J. Sorensen ISBN 3-527-29231-4
- Nicolaou, KC; Yang, Z; Liu, JJ; Ueno, H; Nantermet, PG; Guy, RK; Claiborne, CF; Renaud, J; et al. (February 1994). "Total synthesis of taxol". Nature. 367 (6464): 630–4. Bibcode:1994Natur.367..630N. doi:10.1038/367630a0. PMID 7906395.
- K. C. Nicolaou; P. G. Nantermet; H. Ueno; R. K. Guy; E. A. Couladouros & E. J. Sorensen (1995). "Total Synthesis of Taxol. 1. Retrosynthesis, Degradation, and Reconstitution". J. Am. Chem. Soc. 117 (2): 624–633. doi:10.1021/ja00107a006.
- K. C. Nicolaou; J.-J. Liu; Z. Yang; H. Ueno; E. J. Sorensen; C. F. Claiborne; R. K. Guy; C.-K. Hwang; M. Nakada & P. G. Nantermet (1995). "Total Synthesis of taxol. 2. Construction of A and C ring intermediates and initial attempts to construct the ABC ring system". J. Am. Chem. Soc. 117 (2): 634–644. doi:10.1021/ja00107a007.
- K. C. Nicolaou; Z. Yang; J.-J. Liu; P. G. Nantermet; C. F. Claiborne; J. Renaud; R. K. Guy & K. Shibayama (1995). "Total Synthesis of Taxol. 3. Formation of Taxol's ABC Ring Skeleton". J. Am. Chem. Soc. 117 (2): 645–652. doi:10.1021/ja00107a008.
- K. C. Nicolaou; H. Ueno; J.-J. Liu; P. G. Nantermet; Z. Yang; J. Renaud; K. Paulvannan & R. Chadha (1995). "Total Synthesis of Taxol. 4. The Final Stages and Completion of the Synthesis". J. Am. Chem. Soc. 117 (2): 653–659. doi:10.1021/ja00107a009.