UDP glucuronosyltransferase 1 family, polypeptide A1
UDP-glucuronosyltransferase 1-1 also known as UGT-1A is an enzyme that in humans is encoded by the UGT1A1 gene.[5][6]




UGT-1A is a uridine diphosphate glucuronosyltransferase (UDP-glucuronosyltransferase, UDPGT), an enzyme of the glucuronidation pathway that transforms small lipophilic molecules, such as steroids, bilirubin, hormones, and drugs, into water-soluble, excretable metabolites.[7]
Gene
The UGT1A1 gene is part of a complex locus that encodes several UDP-glucuronosyltransferases. The locus includes thirteen unique alternate first exons followed by four common exons. Four of the alternate first exons are considered pseudogenes. Each of the remaining nine 5' exons may be spliced to the four common exons, resulting in nine proteins with different N-termini and identical C-termini. Each first exon encodes the substrate binding site, and is regulated by its own promoter.[7] Over 100 genetic variants within the UGT1A1 gene have been described, some of which confer increased, reduced or inactive enzymatic activity. The UGT nomenclature committee has compiled a list of these variants, naming each with a * symbol followed by a number.
Clinical significance
Mutations in this gene cause serious problems for bilirubin metabolism; each syndrome can be caused by one or many mutations, so they are differentiated mostly by symptoms and not particular mutations:[8]
- Gilbert syndrome (GS) can be caused by a variety of genetic changes, but in Caucasian and African-American populations, it is most commonly associated with the UGT1A1*28 allele (rs8175347), a homozygous 2-bp insertion (TA) mutation of the TATA box promoter region of the UGT1A1 gene.[8][9][10] This polymorphism impairs proper transcription of UGT1A1 gene, resulting in decreased transcriptional activity of UGT1A1 by about 70%; the resulting reduced enzyme activity leads to the hyperbilirubinemia characteristic of GS.[8][9][11] The *28 polymorphism occurs with a frequency of 26-31% in Caucasians and 42-56% of African-Americans.[12] About 10-15% of these populations are homozygous for the *28 allele, but only 5% actually develop UGT1A1-associated hyperbilirubinemia, so it appears that this mutation alone may be a necessary but not sufficient factor in GS, perhaps acting in combination with other UGT1A1 mutation(s) to increase the chances of developing GS.[8][9] In Asian and Pacific Islander populations, UGT1A1*28 is much less common, occurring at a frequency of approximately 9-16% in Asian populations and 4% of Pacific Islanders.[12][13] In these populations, Gilbert's syndrome is more often due to missense mutations in the coding region of the gene, such as UGT1A1*6 (glycine to arginine substitution at position 71 (G71R); rs4148323) [8][9] A special phenobarbital-responsive enhancer module NR3 region (gtPBREM NR3) helps to increase UDPGT enzyme production, which would make it conceptually possible to medically control the bilirubin level, although this is rarely necessary, particularly in adults (usually the level of total serum bilirubin in Gilbert syndrome patients vary from 1 to 6 mg/dL).[8][9]
- Crigler–Najjar syndrome, type I is associated with mutation(s) that result in a complete absence of normal UGT1A1 enzyme, which causes a severe hyperbilirubinemia with levels of total serum bilirubin from 20 to 45 mg/dL. Phenobarbital treatment does not help to lower bilirubin level, because it only increases the amount of mutated UGT1A1 enzyme, which is still unable to catalyze the glucuronidation of bilirubin, which on the other hand makes phenobarbital treatment diagnostically relevant.[8][14]
- Crigler–Najjar syndrome, type II is associated with other mutation(s) that lead to a reduced activity of the mutated UGT1A1 enzyme, which causes a hyperbilirubinemia with levels of total serum bilirubin from 6 to 20 mg/dL. In this case phenobarbital treatment helps to lower bilirubin lever by more than 30%.[8][15]
- Hyperbilirubinemia, familial transient neonatal (also called breastfeeding jaundice) is associated with mutation(s) that alone do not lead to bilirubin level increase in female patients, but their children when breastfed develop from mild to severe hyperbilirubinemia by receiving steroidal substances (with milk) inhibiting glucuronidation of unconjugated bilirubin that may lead to jaundice and even kernicterus.[8][16]
Pharmacogenetics
Genetic variations within the UGT1A1 gene have also been associated with the development of certain drug toxicities. The UGT1A1*28 variant, the same allele behind many cases of Gilbert syndrome, has been associated with an increased risk for neutropenia in patients receiving the chemotherapeutic drug irinotecan,[17][18] and the U.S. Food and Drug Administration recommends on the irinotecan drug label that patients with the *28/*28 genotype receive a lower starting dose of the drug.[19] The *28 allele has also shown associations with an increased risk for developing diarrhea in patients receiving irinotecan.[17][18] The UGT1A1*6 variant, more common in Asian populations than the *28 variant, has also shown associations with the development of irinotecan toxicities. Patients who are heterozygous or homozygous for the *6 allele may have a higher risk for developing neutropenia and diarrhea as compared to those with the UGT1A1*1/*1 genotype.[17][18]
Interactive pathway map
Click on genes, proteins and metabolites below to link to respective articles. [§ 1]
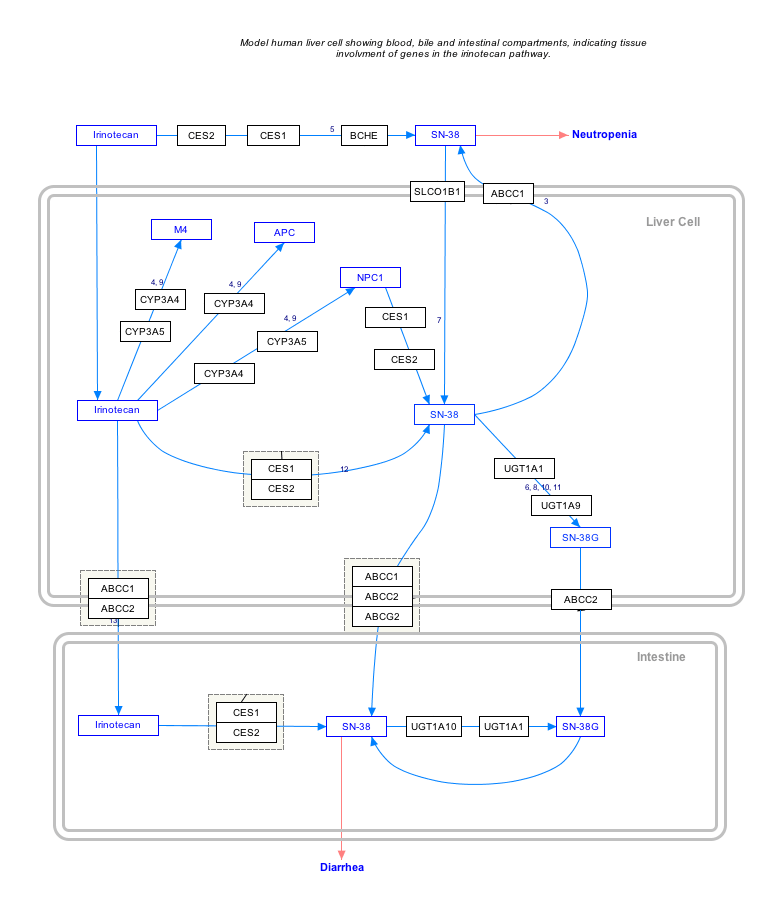
- The interactive pathway map can be edited at WikiPathways: "IrinotecanPathway_WP46359".
See also
- Glucuronosyltransferase
- Lucey-Driscoll syndrome
- Neonatal jaundice
- Cancer pharmacogenomics
References
- GRCh38: Ensembl release 89: ENSG00000241635 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000089960 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Mackenzie PI, Owens IS, Burchell B, Bock KW, Bairoch A, Bélanger A, Fournel-Gigleux S, Green M, Hum DW, Iyanagi T, Lancet D, Louisot P, Magdalou J, Chowdhury JR, Ritter JK, Schachter H, Tephly TR, Tipton KF, Nebert DW (August 1997). "The UDP glycosyltransferase gene superfamily: recommended nomenclature update based on evolutionary divergence". Pharmacogenetics. 7 (4): 255–69. doi:10.1097/00008571-199708000-00001. PMID 9295054.
- Strassburg CP, Manns MP, Tukey RH (April 1998). "Expression of the UDP-glucuronosyltransferase 1A locus in human colon. Identification and characterization of the novel extrahepatic UGT1A8". J. Biol. Chem. 273 (15): 8719–26. doi:10.1074/jbc.273.15.8719. PMID 9535849.
- "Entrez Gene: UGT1A1 UDP glucuronosyltransferase 1 family, polypeptide A1".
- Online Mendelian Inheritance in Man (OMIM): UDP-glycosyltransferase 1 family, polypeptide A1; UGT1A1 - 191740
- Online Mendelian Inheritance in Man (OMIM): Gilbert syndrome - 143500
- Beutler E, Gelbart T, Demina A (July 1998). "Racial variability in the UDP-glucuronosyltransferase 1 (UGT1A1) promoter: a balanced polymorphism for regulation of bilirubin metabolism?". Proc Natl Acad Sci USA. 95 (14): 8170–4. Bibcode:1998PNAS...95.8170B. doi:10.1073/pnas.95.14.8170. PMC 20948. PMID 9653159.
- Tukey RH, Strassburg CP, Mackenzie PI (September 2002). "Pharmacogenomics of human UDP-glucuronosyltransferases and irinotecan toxicity". Mol Pharmacol. 62 (3): 446–50. doi:10.1124/mol.62.3.446. PMID 12181419.
- Barbarino JM, Haidar CE, Klein TE, Altman RB (March 2014). "PharmGKB summary: very important pharmacogene information for UGT1A1". Pharmacogenet Genomics. 24 (3): 177–83. doi:10.1097/FPC.0000000000000024. PMC 4091838. PMID 24492252.
- AlFadhli S, Al-Jafer H, Hadi M, Al-Mutairi M, Nizam R (October 2013). "The effect of UGT1A1 promoter polymorphism in the development of hyperbilirubinemia and cholelithiasis in hemoglobinopathy patients". PLOS ONE. 8 (10): e77681. Bibcode:2013PLoSO...877681A. doi:10.1371/journal.pone.0077681. PMC 3813713. PMID 24204915.
- Online Mendelian Inheritance in Man (OMIM): Crigler–Najjar syndrome, type I - 218800
- Online Mendelian Inheritance in Man (OMIM): Crigler–Najjar syndrome, type II - 606785
- Online Mendelian Inheritance in Man (OMIM): Hyperbilirubinemia, transient familial neonatal - 237900
- Marsh S, Hoskins JM (July 2010). "Irinotecan pharmacogenomics". Pharmacogenomics. 11 (7): 1003–10. doi:10.2217/pgs.10.95. PMC 2927346. PMID 20602618.
- Barbarino JM, Haidar CE, Klein TE, Altman RB (March 2014). "PharmGKB summary: very important pharmacogene information for UGT1A1". Pharmacogenetics and Genomics. 24 (3): 177–83. doi:10.1097/fpc.0000000000000024. PMC 4091838. PMID 24492252.
- "CAMPTOSAR (irinotecan hydrochloride injection, solution) drug label". DailyMed. U.S. National Library of Medicine. Retrieved 5 January 2015.
Further reading
- Tukey RH, Strassburg CP (2000). "Human UDP-glucuronosyltransferases: metabolism, expression, and disease". Annu. Rev. Pharmacol. Toxicol. 40: 581–616. doi:10.1146/annurev.pharmtox.40.1.581. PMID 10836148.
- Kadakol A, Ghosh SS, Sappal BS, Sharma G, Chowdhury JR, Chowdhury NR (2000). "Genetic lesions of bilirubin uridine-diphosphoglucuronate glucuronosyltransferase (UGT1A1) causing Crigler-Najjar and Gilbert syndromes: correlation of genotype to phenotype". Hum. Mutat. 16 (4): 297–306. doi:10.1002/1098-1004(200010)16:4<297::AID-HUMU2>3.0.CO;2-Z. PMID 11013440.
- King CD, Rios GR, Green MD, Tephly TR (2001). "UDP-glucuronosyltransferases". Curr. Drug Metab. 1 (2): 143–61. doi:10.2174/1389200003339171. PMID 11465080.
- Bosma PJ (2003). "Inherited disorders of bilirubin metabolism". J. Hepatol. 38 (1): 107–17. doi:10.1016/S0168-8278(02)00359-8. PMID 12480568.
- Innocenti F, Ratain MJ (2003). "Irinotecan treatment in cancer patients with UGT1A1 polymorphisms". Oncology (Williston Park, N.Y.). 17 (5 Suppl 5): 52–5. PMID 12800608.
- Lee W, Lockhart AC, Kim RB, Rothenberg ML (2005). "Cancer pharmacogenomics: powerful tools in cancer chemotherapy and drug development". Oncologist. 10 (2): 104–11. doi:10.1634/theoncologist.10-2-104. PMID 15709212.
- Navarro SL, Peterson S, Chen C, Makar KW, Schwarz Y, King IB, Li SS, Li L, Kestin M, Lampe JW (2009). "Cruciferous vegetable feeding alters UGT1A1 activity: diet and genotype-dependent changes in serum bilirubin in a controlled trial". Cancer Prev. Res. 2 (4): 345–52. doi:10.1158/1940-6207.CAPR-08-0178. PMC 2666928. PMID 19336732.
- Barbarino JM, Haidar CE, Klein TE, Altman RB (March 2014). "PharmGKB summary: very important pharmacogene information for UGT1A1". Pharmacogenet Genomics. 24 (3): 177–83. doi:10.1097/FPC.0000000000000024. PMC 4091838. PMID 24492252.
External links
- UGT1A1+protein,+human at the US National Library of Medicine Medical Subject Headings (MeSH)
- UGT nomenclature homepage
- PharmGKB page for UGT1A1