Kaolinite
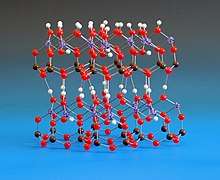
Kaolinite (/ˈkeɪəlɪnaɪt/)[4][5] is a clay mineral, part of the group of industrial minerals with the chemical composition Al2Si2O5(OH)4. It is a layered silicate mineral, with one tetrahedral sheet of silica (SiO
4) linked through oxygen atoms to one octahedral sheet of alumina (AlO
6) octahedra.[6] Rocks that are rich in kaolinite are known as kaolin /ˈkeɪəlɪn/ or china clay.[7]
| Kaolinite | |
|---|---|
 | |
| General | |
| Category | Phyllosilicates Kaolinite-serpentine group |
| Formula (repeating unit) | Al 2(OH) 4Si 2O 5 |
| Strunz classification | 9.ED.05 |
| Crystal system | Triclinic |
| Crystal class | Pedial (1) (same H-M symbol) |
| Space group | P1 |
| Unit cell | a = 5.13 Å, b = 8.89 Å c = 7.25 Å; α = 90° β = 104.5°, γ = 89.8°; Z = 2 |
| Identification | |
| Color | White to cream, sometimes red, blue or brown tints from impurities and pale-yellow; also often stained various hues, tans and browns being common. |
| Crystal habit | Rarely as crystals, thin plates or stacked, More commonly as microscopic pseudohexagonal plates and clusters of plates, aggregated into compact, claylike masses |
| Cleavage | Perfect on {001} |
| Tenacity | Flexible but inelastic |
| Mohs scale hardness | 2–2.5 |
| Luster | Pearly to dull earthy |
| Streak | White |
| Specific gravity | 2.16–2.68 |
| Optical properties | Biaxial (–) |
| Refractive index | nα = 1.553–1.565, nβ = 1.559–1.569, nγ = 1.569–1.570 |
| 2V angle | Measured: 24° to 50°, Calculated: 44° |
| References | [1][2][3] |
| Kaolinite | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Traditional Chinese | 高嶺石 | ||||||||||
| Simplified Chinese | 高岭石 | ||||||||||
| Literal meaning | "Gaoling stone" | ||||||||||
| |||||||||||
The name "kaolin" is derived from "Gaoling" (Chinese: 高嶺; pinyin: Gāolǐng; lit.: 'High Ridge'), a Chinese village near Jingdezhen in southeastern China's Jiangxi Province.[8] The name entered English in 1727 from the French version of the word: kaolin, following François Xavier d'Entrecolles's reports on the making of Jingdezhen porcelain.[9]
Kaolinite has a low shrink–swell capacity and a low cation-exchange capacity (1–15 meq/100 g). It is a soft, earthy, usually white, mineral (dioctahedral phyllosilicate clay), produced by the chemical weathering of aluminium silicate minerals like feldspar. In many parts of the world it is colored pink-orange-red by iron oxide, giving it a distinct rust hue. Lighter concentrations yield white, yellow, or light orange colors. Alternating layers are sometimes found, as at Providence Canyon State Park in Georgia, United States. Commercial grades of kaolin are supplied and transported as dry powder, semi-dry noodle, or liquid slurry.
Chemistry
Notation
The chemical formula for kaolinite as used in mineralogy is Al
2Si
2O
5(OH)
4,[3] however, in ceramics applications the formula is typically written in terms of oxides, thus the formula for kaolinite is Al
2O
3·2SiO
2·2H2O.[10]
Structural transformations
Kaolinite group clays undergo a series of phase transformations upon thermal treatment in air at atmospheric pressure.
Drying
Below 100 °C (212 °F), exposure to dry air will slowly remove liquid water from the kaolin. The end-state for this transformation is referred to as "leather dry". Between 100 °C and about 550 °C (1,022 °F), any remaining liquid water is expelled from kaolinite. The end state for this transformation is referred to as "bone dry". Throughout this temperature range, the expulsion of water is reversible: if the kaolin is exposed to liquid water, it will be reabsorbed and disintegrate into its fine particulate form. Subsequent transformations are not reversible, and represent permanent chemical changes.
Metakaolin
Endothermic dehydration of kaolinite begins at 550–600 °C producing disordered metakaolin, but continuous hydroxyl loss is observed up to 900 °C (1,650 °F).[11] Although historically there was much disagreement concerning the nature of the metakaolin phase, extensive research has led to a general consensus that metakaolin is not a simple mixture of amorphous silica (SiO
2) and alumina (Al
2O
3), but rather a complex amorphous structure that retains some longer-range order (but not strictly crystalline) due to stacking of its hexagonal layers.[11]

Spinel
Further heating to 925–950 °C converts metakaolin to an aluminium-silicon spinel which is sometimes also referred to as a gamma-alumina type structure:

Platelet mullite
Upon calcination above 1050 °C, the spinel phase nucleates and transforms to platelet mullite and highly crystalline cristobalite:

Occurrence
Kaolinite is one of the most common minerals; it is mined, as kaolin, in Malaysia, Pakistan, Vietnam, Brazil, Bulgaria, Bangladesh, France, the United Kingdom, Iran, Germany, India, Australia, South Korea, the People's Republic of China, the Czech Republic, Spain, South Africa, and the United States.[1]
Mantles of kaolinitic saprolite are common in Western and Northern Europe. The ages of these mantles are Mesozoic to Early Cenozoic.[12]
Kaolinite clay occurs in abundance in soils that have formed from the chemical weathering of rocks in hot, moist climates—for example in tropical rainforest areas. Comparing soils along a gradient towards progressively cooler or drier climates, the proportion of kaolinite decreases, while the proportion of other clay minerals such as illite (in cooler climates) or smectite (in drier climates) increases. Such climatically-related differences in clay mineral content are often used to infer changes in climates in the geological past, where ancient soils have been buried and preserved.
In the Institut National pour l'Etude Agronomique au Congo Belge (INEAC) classification system, soils in which the clay fraction is predominantly kaolinite are called kaolisol (from kaolin and soil).[13]
In the US, the main kaolin deposits are found in central Georgia, on a stretch of the Atlantic Seaboard fall line between Augusta and Macon. This area of thirteen counties is called the "white gold" belt; Sandersville is known as the "Kaolin Capital of the World" due to its abundance of kaolin.[14][15] In the late 1800s, an active kaolin surface-mining industry existed in the extreme southeast corner of Pennsylvania, near the towns of Landenberg and Kaolin, and in what is present-day White Clay Creek Preserve. The product was brought by train to Newark, Delaware, on the Newark-Pomeroy line, along which can still be seen many open-pit clay mines. The deposits were formed between the late Cretaceous and early Paleogene, about 100 to 45 million years ago, in sediments derived from weathered igneous and metakaolin rocks.[8] Kaolin production in the US during 2011 was 5.5 million tons.[16]
During the Paleocene–Eocene Thermal Maximum sediments were enriched with kaolinite from a detrital source due to denudation.[17]
Synthesis and genesis
Difficulties are encountered when trying to explain kaolinite formation under atmospheric conditions by extrapolation of thermodynamic data from the more successful high-temperature syntheses (as for example Meijer and Van der Plas, 1980[18] have pointed out). La Iglesia and Van Oosterwijk-Gastuche (1978)[19] thought that the conditions under which kaolinite will nucleate can be deduced from stability diagrams, based as they are on dissolution data. Because of a lack of convincing results in their own experiments, La Iglesia and Van Oosterwijk-Gastuche (1978) had to conclude, however, that there were other, still unknown, factors involved in the low-temperature nucleation of kaolinite. Because of the observed very slow crystallization rates of kaolinite from solution at room temperature Fripiat and Herbillon (1971) postulated the existence of high activation energies in the low-temperature nucleation of kaolinite.
At high temperatures, equilibrium thermodynamic models appear to be satisfactory for the description of kaolinite dissolution and nucleation, because the thermal energy suffices to overcome the energy barriers involved in the nucleation process. The importance of syntheses at ambient temperature and atmospheric pressure towards the understanding of the mechanism involved in the nucleation of clay minerals lies in overcoming these energy barriers. As indicated by Caillère and Hénin (1960)[20] the processes involved will have to be studied in well-defined experiments, because it is virtually impossible to isolate the factors involved by mere deduction from complex natural physico-chemical systems such as the soil environment. Fripiat and Herbillon (1971),[21] in a review on the formation of kaolinite, raised the fundamental question how a disordered material (i.e., the amorphous fraction of tropical soils) could ever be transformed into a corresponding ordered structure. This transformation seems to take place in soils without major changes in the environment, in a relatively short period of time, and at ambient temperature (and pressure).
Low-temperature synthesis of clay minerals (with kaolinite as an example) has several aspects. In the first place the silicic acid to be supplied to the growing crystal must be in a monomeric form, i.e., silica should be present in very dilute solution (Caillère et al., 1957;[22] Caillère and Hénin, 1960;[20] Wey and Siffert, 1962;[23] Millot, 1970[24]). In order to prevent the formation of amorphous silica gels precipitating from supersaturated solutions without reacting with the aluminium or magnesium cations to form crystalline silicates, the silicic acid must be present in concentrations below the maximum solubility of amorphous silica. The principle behind this prerequisite can be found in structural chemistry: "Since the polysilicate ions are not of uniform size, they cannot arrange themselves along with the metal ions into a regular crystal lattice." (Iler, 1955, p. 182[25])
The second aspect of the low-temperature synthesis of kaolinite is that the aluminium cations must be hexacoordinated with respect to oxygen (Caillère and Hénin, 1947;[26] Caillère et al., 1953;[27] Hénin and Robichet, 1955[28]). Gastuche et al. (1962),[29] as well as Caillère and Hénin (1962) have concluded, that only in those instances when the aluminium hydroxide is in the form of gibbsite, kaolinite can ever be formed. If not, the precipitate formed will be a "mixed alumino-silicic gel" (as Millot, 1970, p. 343 put it). If it were the only requirement, large amounts of kaolinite could be harvested simply by adding gibbsite powder to a silica solution. Undoubtedly a marked degree of adsorption of the silica in solution by the gibbsite surfaces will take place, but, as stated before, mere adsorption does not create the layer lattice typical of kaolinite crystals.
The third aspect is that these two initial components must be incorporated into one and the same mixed crystal with a layer structure. From the following equation (as given by Gastuche and DeKimpe, 1962)[30] for kaolinite formation

it can be seen, that five molecules of water must be removed from the reaction for every molecule of kaolinite formed. Field evidence illustrating the importance of the removal of water from the kaolinite reaction has been supplied by Gastuche and DeKimpe (1962). While studying soil formation on a basaltic rock in Kivu (Zaïre), they noted how the occurrence of kaolinite depended on the "degrée de drainage" of the area involved. A clear distinction was found between areas with good drainage (i.e., areas with a marked difference between wet and dry seasons) and those areas with poor drainage (i.e., perennially swampy areas). Only in the areas with distinct seasonal alternations between wet and dry was kaolinite found. The possible significance of alternating wet and dry conditions on the transition of allophane into kaolinite has been stressed by Tamura and Jackson (1953).[31] The role of alternations between wetting and drying on the formation of kaolinite has also been noted by Moore (1964).[32]
Laboratory syntheses
Syntheses of kaolinite at high temperatures (more than 100 °C [212 °F]) are relatively well known. There are for example the syntheses of Van Nieuwenberg and Pieters (1929);[33] Noll (1934);[34] Noll (1936);[35] Norton (1939);[36] Roy and Osborn (1954);[37] Roy (1961);[38] Hawkins and Roy (1962);[39] Tomura et al. (1985);[40] Satokawa et al. (1994)[41] and Huertas et al. (1999).[42] Relatively few low-temperature syntheses have become known (cf. Brindley and DeKimpe (1961);[43] DeKimpe (1969);[44] Bogatyrev et al. (1997)[45]).
Laboratory syntheses of kaolinite at room temperature and atmospheric pressure have been described by DeKimpe et al. (1961).[46] From those tests the role of periodicity becomes convincingly clear. DeKimpe et al. (1961) had used daily additions of alumina (as AlCl
3·6 H
2O) and silica (in the form of ethyl silicate) during at least two months. In addition, adjustments of the pH took place every day by way of adding either hydrochloric acid or sodium hydroxide. Such daily additions of Si and Al to the solution in combination with the daily titrations with hydrochloric acid or sodium hydroxide during at least 60 days will have introduced the necessary element of periodicity.
Only now the actual role of what has been described as the "aging" (Alterung) of amorphous alumino-silicates (as for example Harder, 1978[47] had noted) can be fully understood. Time as such is not bringing about any change in a closed system at equilibrium; but a series of alternations, of periodically changing conditions (by definition, taking place in an open system), will bring about the low-temperature formation of more and more of the stable phase kaolinite instead of (ill-defined) amorphous alumino-silicates.
Uses
The main use of the mineral kaolinite (about 50% of the time) is the production of paper; its use ensures the gloss on some grades of coated paper.[48]
Kaolin is also known for its capabilities to induce and accelerate blood clotting. In April 2008 the US Naval Medical Research Institute announced the successful use of a kaolinite-derived aluminosilicate infusion in traditional gauze, known commercially as QuikClot Combat Gauze,[49] which is still the hemostat of choice for all branches of the US military.
Kaolin is used (or was used in the past):
- in ceramics (it is the main component of porcelain)
- in toothpaste
- as a light-diffusing material in white incandescent light bulbs
- in cosmetics
- in industrial insulation material called Kaowool (a form of mineral wool)
- in 'pre-work' skin protection and barrier creams[50]
- in paint to extend the titanium dioxide (TiO
2) white pigment and modify gloss levels - for modifying the properties of rubber upon vulcanization
- in adhesives to modify rheology[51]
- in organic farming as a spray applied to crops to deter insect damage, and in the case of apples, to prevent sun scald
- as whitewash in traditional stone masonry homes in Nepal (the most common method is to paint the upper part with white kaolin clay and the middle with red clay; the red clay may extend to the bottom, or the bottom may be painted black)
- as a filler in Edison Diamond Discs[52]
- as a filler to give bulk, or a coating to improve the surface in papermaking
- as an indicator in radiological dating since kaolinite can contain very small traces of uranium and thorium
- to soothe an upset stomach, similar to the way parrots (and later, humans) in South America originally used it[53] (more recently, industrially-produced kaolinite preparations were common for treatment of diarrhea; the most common of these was Kaopectate, which abandoned the use of kaolin in favor of attapulgite and then (in the United States) bismuth subsalicylate (the active ingredient in Pepto-Bismol))
- for facial masks or soap (known as "White Clay")
- as adsorbents in water and wastewater treatment[54]
- to induce blood clotting in diagnostic procedures, e.g. Kaolin clotting time
- in its altered metakaolin form, as a pozzolan; when added to a concrete mix, metakaolin accelerates the hydration of Portland cement and takes part in the pozzolanic reaction with the portlandite formed in the hydration of the main cement minerals (e.g. alite)
- in its altered metakaolin form, as a base component for geopolymer compounds
Geophagy
Humans sometimes eat kaolin for health or to suppress hunger,[55] a practice known as geophagy. Consumption is greater among women, especially during pregnancy.[56] This practice has also been observed within a small population of African-American women in the Southern United States, especially Georgia.[57][58] There, the kaolin is called white dirt, chalk, or white clay.[57]
Safety
People can be exposed to kaolin in the workplace by breathing in the powder or from skin or eye contact.
United States
The Occupational Safety and Health Administration (OSHA) has set the legal limit (permissible exposure limit) for kaolin exposure in the workplace as 15 mg/m3 total exposure and 5 mg/m3 respiratory exposure over an 8-hour workday. The National Institute for Occupational Safety and Health (NIOSH) has set a recommended exposure limit (REL) of 10 mg/m3 total exposure TWA 5 mg/m3 respiratory exposure over an 8-hour workday.[59]
See also
- China stone
- Clay pit
- Dickite
- Halloysite – Aluminosilicate clay mineral
- Kaolin Deposits of Charentes Basin, France
- Kaolin spray
- Medicinal clay
- Nacrite
References
Citations
- "Kaolinite mineral information and data". MinDat.org. Retrieved 5 August 2009.
- "Kaolinite Mineral Data". McDougall Minerals. Retrieved 5 August 2009.
- Anthony JW, Bideaux RA, Bladh KW, et al., eds. (1995). "Kaolinite" (PDF). Handbook of Mineralogy: Silica, silicates. Tucson, Ariz.: Mineral Data Publishing. ISBN 9780962209734. OCLC 928816381.
- "kaolinite". Dictionary.com Unabridged. Random House.
- "Kaolinite". Oxford Dictionaries (online). 20 January 2016.
- Deer WA, Howie RA, Zussman J (1992). An Introduction to the Rock-forming Minerals (2nd ed.). Harlow: Longman. ISBN 9780470218099.
- Pohl WL (2011). Economic geology: principles and practice: metals, minerals, coal and hydrocarbons – introduction to formation and sustainable exploitation of mineral deposits. Chichester, West Sussex: Wiley-Blackwell. p. 331. ISBN 9781444336627.
- Schroeder PA (31 July 2018). "Kaolin". New Georgia Encyclopedia (online). Retrieved 14 March 2019.
- Harper, Douglas. "kaolin". Online Etymology Dictionary.
- Perry DL (2011). Handbook of Inorganic Compounds (2nd ed.). Boca Raton: Taylor & Francis. ISBN 9781439814611. OCLC 587104373.
- Bellotto M, Gualtieri A, Artioli G, et al. (1995). "Kinetic study of the kaolinite-mullite reaction sequence. Part I: kaolinite dehydroxylation". Phys. Chem. Miner. 22 (4): 207–214. Bibcode:1995PCM....22..207B. doi:10.1007/BF00202253.
- Migoń P, Lidmar-Bergström K (2002). "Deep weathering through time in central and northwestern Europe: problems of dating and interpretation of geological record". Catena. 49 (1–2): 25–40. doi:10.1016/S0341-8162(02)00015-2.
- Young A (1980). Tropical soils and soil survey. Cambridge Geographical Studies. 9. CUP Archive. p. 132. ISBN 9780521297684.
- "Kaolin Capital of the World". City of Sandersville, GA. Retrieved 27 August 2018.
- Reece C. "Making Peace With the Age-Old Practice of Eating White Dirt". The Bitter Southerner. Retrieved 27 August 2018.
- Virta R (2012). Mineral Commodity Summaries (PDF) (Technical report). U.S. Geological Survey. pp. 44–45.
- Adatte T, Khozyem H, Spangenberg JE, et al. (2014). "Response of terrestrial environment to the Paleocene-Eocene Thermal Maximum (PETM), new insights from India and NE Spain". Rendiconti Online della Società Geologica Italiana. 31: 5–6. doi:10.3301/ROL.2014.17.
- Meijer EL, van der Plas L (1980). Relative stabilities of soil minerals. Mededelingen Landbouwhogeschool Wageningen. 80. Wageningen: Veenman. p. 18.
- La Iglesia A, Van Oosterwyck-Gastuche MC (1978). "Kaolinite Synthesis. I. Crystallization Conditions at Low Temperatures and Calculation of Thermodynamic Equilibria. Application to Laboratory and Field Observations". Clays and Clay Minerals. 26 (6): 397–408. Bibcode:1978CCM....26..397L. doi:10.1346/CCMN.1978.0260603.
- Caillère S, Hénin S (1960). "Vues d'ensemble sur le problème de la synthèse des minéraux argileux à basse température". Bulletin du Groupe français des argiles (in French). 12 (7): 63. doi:10.3406/argil.1960.969.
- Fripiat JJ, Herbillon AJ (1971). "Formation and transformations of clay minerals in tropical soils". Soils and tropical weathering: proceedings of the Bandung Symposium 16 to 23 November 1969. Natural resources research. 11. Paris: Unesco. pp. 15–24. OCLC 421565.
- Caillère S, Hénin S, Esquevin J (1957). "Synthèse des minéraux argileux". Bulletin du Groupe français des argiles (in French). 9 (4): 67–76. doi:10.3406/argil.1957.940.
- Wey R, Siffert B (1961). "Réactions de la silice monomoléculaire en solutions avec les ions Al3+ et Mg2+". Colloques Internationaux (in French). Centre National des Recherches Scientifiques. 105: 11–23.
- Millot G (1970). Geology of Clays. Translated by Paquet H, Farrand WR. New York: Springer-Verlag. doi:10.1007/978-3-662-41609-9. ISBN 9783662416099.
- Iler RK (1955). The colloid chemistry of silica and silicates. Ithaca, N.Y.: Cornell University Press.
- Caillère S, Hénin S (1947). "Formation d'une phyllite du type kaolinique par traitement d'une montmorillonite". Comptes Rendus de l'Académie des Sciences de Paris. 224 (1): 53–55.
- Caillère S, Hénin S, Esquevin J (1953). "Recherches sur la synthèse des minéraux argileux". Bulletin de la Société française de Minéralogie et de Cristallographie (in French). 76 (7): 300–314. doi:10.3406/bulmi.1953.4841.
- Hénin S, Robichet O (1955). "Résultats obtenus au cours de nouveaux essais de synthèse de minéraux argileux". Bulletin du Groupe français des argiles (in French). 6 (1): 19–22. doi:10.3406/argil.1955.1257.
- Gastuche MC, Fripiat JJ, DeKimpe C (1962). "La genèse des minéraux argileux de la famille du kaolin. I. – Aspect colloidal". Colloque C.N.R.S. 105: 57–65.
- Gastuche MC, DeKimpe C (1962). "La genèse des minéraux argileux de la famille du kaolin. II. Aspect cristallin". Colloque C.N.R.S. 105: 75–88.
- Tamura T, Jackson ML (1953). "Structural and Energy Relationships in the Formation of Iron and Aluminum Oxides, Hydroxides, and Silicates". Science. 117 (3041): 381–383. Bibcode:1953Sci...117..381T. doi:10.1126/science.117.3041.381. PMID 17749950.
- Moore LR (1964). "The in Situ Formation and Development of Some Kaolinite Macrocrystals". Clay Minerals. 5 (31): 338–352. Bibcode:1964ClMin...5..338M. doi:10.1180/claymin.1964.005.31.02.
- van Nieuwenburg CJ, Pieters HA (1929). "Studies on hydrated aluminium silicates: I. The rehydration of metakaolin and the synthesis of kaolin". Recl. Trav. Chim. Pays-Bas. 48 (1): 27–36. doi:10.1002/recl.19290480106.
- Noll W (1934). "Hydrothermale Synthese des Kaolins". Zeitschrift für Kristallographie, Mineralogie und Petrographie (in German). 45 (2–3): 175–190. Bibcode:1934ZKMP...45..175N. doi:10.1007/BF02943371.
- Noll W (1936). "Über die Bildungsbedingungen von Kaolin, Montmorillonit, Sericit, Pyrophyllit und Analcim". Zeitschrift für Kristallographie, Mineralogie und Petrographie (in German). 48 (3–4): 210–247. Bibcode:1936ZKMP...48..210N. doi:10.1007/BF02939458.
- Norton FH (1939). "Hydrothermal formation of clay minerals in the laboratory". Am. Mineral. 24 (1): 1–17.
- Roy R, Osborn EF (1954). "The system Al
2O
3-SiO
2-H
2O". Am. Mineral. 39 (11–12): 853–885. - Roy R (1962). "The preparation and properties of synthetic clay minerals". Colloque C.N.R.S. 105: 83–98.
- Hawkins DB, Roy R (1962). "Electrolytic Synthesis of Kaolinite Under Hydrothermal Conditions". J. Am. Ceram. Soc. 45 (10): 507–508. doi:10.1111/j.1151-2916.1962.tb11044.x.
- Tomura S, Shibasaki Y, Mizuta H, et al. (1985). "Growth Conditions and Genesis of Spherical and Platy Kaolinite". Clays and Clay Minerals. 33 (3): 200–206. Bibcode:1985CCM....33..200T. doi:10.1346/CCMN.1985.0330305.
- Satokawa S, Osaki Y, Samejima S, et al. (1994). "Effects of the Structure of Silica-Alumina Gel on the Hydrothermal Synthesis of Kaolinite". Clays and Clay Minerals. 42 (3): 288–297. Bibcode:1994CCM....42..288S. doi:10.1346/CCMN.1994.0420307.
- Huertas FJ, Fiore S, Huertas F, et al. (1999). "Experimental study of the hydrothermal formation of kaolinite". Chemical Geology. 156 (1–4): 171–190. Bibcode:1999ChGeo.156..171H. doi:10.1016/S0009-2541(98)00180-6.
- Brindley GW, De Kimpe C (1961). "Attempted Low-Temperature Syntheses of Kaolin Minerals". Nature. 190 (4772): 254. Bibcode:1961Natur.190..254B. doi:10.1038/190254a0.
- De Kimpe CR (1969). "Crystallization of kaolinite at low temperature from an alumino-silicic gel". Clays and Clay Minerals. 17 (1): 37–38. Bibcode:1969CCM....17...37D. doi:10.1346/CCMN.1969.0170107.
- Bogatyrev BA, Mateeva LA, Zhukov VV, et al. (1997). "Low-temperature synthesis of kaolinite and halloysite on the gibbsite – silicic acid solution system". Transactions (Doklady) of the Russian Academy of Sciences. Earth science sections. 353 A: 403–405.
- DeKimpe CR, Gastuche MC, Brindley GW (1961). "Ionic coordination in alumino-silicic acids in relation to clay mineral formation" (PDF). Am. Mineral. 46 (11–12): 1370–1381.
- Harder H (1978). "Synthesen von Tonmineralen unter spezieller Berücksichtigung festländischer Bedingungen". Schriftenreihe für geologische Wissenschaften (Berlin) (in German). 11: 51–78.
- Murray HH, Lyons SC (1955). "Correlation of Paper-Coating Quality with Degree of Crystal Perfection of Kaolinite". Clays and Clay Minerals. 4 (1): 31–40. Bibcode:1955CCM.....4...31M. doi:10.1346/CCMN.1955.0040105.
- Rowe A (24 April 2008). "Nanoparticles Help Gauze Stop Gushing Wounds". Wired. Condé Nast. Archived from the original on 6 July 2009. Retrieved 5 August 2009.
- "Stokoderm® Protect PURE" (PDF). debgroup.com (product leaflet). Deb USA, Inc. 2017. Retrieved 12 April 2018.
- Ciullo PA (1996). Industrial Minerals and Their Uses: A Handbook and Formulary. Westwood, NJ: Noyes Publications. pp. 41–43. ISBN 9780815518082.
- Gracyk T (2006). "Edison Diamond Discs: 1912 - 1929". Tim Gracyk's Phonographs, Singers, & Old Records. Retrieved 22 March 2019.
- Diamond JM (1999). "Dirty eating for healthy living". Nature. Evolutionary biology. 400 (6740): 120–121. Bibcode:1999Natur.400..120D. doi:10.1038/22014. PMID 10408435.
- Leiviskä T, Gehör S, Eijärvi E, et al. (2012). "Characteristics and potential applications of coarse clay fractions from Puolanka, Finland". Open Eng. 2 (2): 239–247. Bibcode:2012CEJE....2..239L. doi:10.2478/s13531-011-0067-9.
- Kamtche F (2012). "Balengou: autour des mines" [Balengou: around the mines]. Le Jour (in French). Archived from the original on 4 March 2012. Retrieved 22 March 2019.
- Callahan GN (2003). "Eating Dirt". Emerg. Infect. Dis. CDC. 9 (8): 1016–1021. doi:10.3201/eid0908.ad0908. PMC 3020602. PMID 12971372.
- Grigsby RK (3 February 2004). "Clay Eating". New Georgia Encyclopedia (online). Science & Medicine. Retrieved 20 October 2019.
- Chen L (2 April 2014). "The Old And Mysterious Practice of Eating Dirt, Revealed". The Salt. NPR.
- "Kaolin". NIOSH Pocket Guide to Chemical Hazards. CDC. Retrieved 6 November 2015.
General references
- Deer WA, Howie RA, Zussman J (1992). An introduction to the rock-forming minerals (2nd ed.). Harlow: Longman. ISBN 0582300940.
- Hurlbut CS, Klein C (1985). Manual of mineralogy – after J. D. Dana (20th ed.). Wiley. pp. 428–429. ISBN 0471805807.
- Breck DW (1984). Zeolite molecular sieves. Malabar, FL: R. E. Krieger Publishing Co. pp. 314–315. ISBN 0898746485.