Cockayne syndrome
Cockayne syndrome (CS), also called Neill-Dingwall syndrome, is a rare and fatal autosomal recessive neurodegenerative disorder characterized by growth failure, impaired development of the nervous system, abnormal sensitivity to sunlight (photosensitivity), eye disorders and premature aging.[1][2][3] Failure to thrive and neurological disorders are criteria for diagnosis, while photosensitivity, hearing loss, eye abnormalities, and cavities are other very common features.[3] Problems with any or all of the internal organs are possible. It is associated with a group of disorders called leukodystrophies, which are conditions characterized by degradation of neurological white matter. The underlying disorder is a defect in a DNA repair mechanism.[4] Unlike other defects of DNA repair, patients with CS are not predisposed to cancer or infection.[5] Cockayne syndrome is a rare but destructive disease usually resulting in death within the first or second decade of life. The mutation of specific genes in Cockayne syndrome is known, but the widespread effects and its relationship with DNA repair is yet to be well understood.[5]
| Cockayne syndrome | |
|---|---|
| Other names | Neill-Dingwall syndrome |
| Specialty | Medical genetics, neurology, dermatology |
It is named after English physician Edward Alfred Cockayne (1880–1956) who first described it in 1936 and re-described in 1946.[6] Neill-Dingwall syndrome was named after Mary M. Dingwall and Catherine A. Neill.[6] These two scientists described the case of two brothers with Cockayne syndrome and asserted it was the same disease described by Cockayne. In their article the two contributed to the signs of the disease through their discovery of calcifications in the brain. They also compared Cockayne syndrome to what is now known as Hutchinson–Gilford progeria syndrome (HGPS), then called progeria, due to the advanced aging that characterizes both disorders.[6]
Types
- CS Type I, the "classic" form, is characterized by normal fetal growth with the onset of abnormalities in the first two years of life. Vision and hearing gradually decline.[7] The central and peripheral nervous systems progressively degenerate until death in the first or second decade of life as a result of serious neurological degradation. Cortical atrophy is less severe in CS Type I.[8]
- CS Type II is present from birth (congenital) and is much more severe than CS Type 1.[7] It involves very little neurological development after birth. Death usually occurs by age seven. This specific type has also been designated as cerebro-oculo-facio-skeletal (COFS) syndrome or Pena-Shokeir syndrome Type II.[7] COFS syndrome is named so due to the effects it has on the brain, eyes, face, and skeletal system, as the disease frequently causes brain atrophy, cataracts, loss of fat in the face, and osteoporosis. COFS syndrome can be further subdivided into several conditions (COFS types 1, 2, 3 (associated with xeroderma pigmentosum) and 4).[9] Typically patients with this early-onset form of the disorder show more severe brain damage, including reduced myelination of white matter, and more widespread calcifications, including in the cortex and basal ganglia.[8]
- CS Type III, characterized by late-onset, is typically milder than Types I and II.[7] Often patients with Type III will live into adulthood.
- Xeroderma pigmentosum-Cockayne syndrome (XP-CS) occurs when an individual also suffers from xeroderma pigmentosum, another DNA repair disease. Some symptoms of each disease are expressed. For instance, freckling and pigment abnormalities characteristic of XP are present. The neurological disorder, spasticity, and underdevelopment of sexual organs characteristic of CS are seen. However, hypomyelination and the facial features of typical CS patients are not present.[10]
Causes
If hyperoxia or excess oxygen occurs in our body, our cellular metabolism produce several highly reactive forms of oxygen called free radicals. This can cause oxidative damage to cellular components including the DNA. In normal cells, our body repairs the damaged sections. In the case of this disease, Due to subtle defects in transcription, children's genetic machinery for synthesizing proteins needed by the body does not operate at normal capacity. That is, scientists believed that these children's genetic machinery for synthesizing proteins needed by the body does not operate at normal capacity. Over time, went this theory, results in developmental failure and death. Every minute, the body pumps 10 to 20 liters of oxygen through the blood, carrying it to billions of cells in our bodies. In its normal molecular form, oxygen is harmless. However, cellular metabolism involving oxygen can generate several highly reactive free radicals. These free radicals can cause oxidative damage to cellular components including the DNA. In an average human cell, several thousand lesions occur in the DNA every day. Many of these lesions result from oxidative damage. Each lesion -- a damaged section of DNA -- must be snipped out and the DNA repaired to preserve its normal function. Unrepaired DNA can lose its ability to code for proteins. Mutations also can result. These mutations can activate oncogenes or silence tumor suppressor genes. According to research, oxidative damage to active genes is not preferentially repaired, and in the most severe cases, the repair is slowed throughout the whole genome. The resulting accumulation of oxidative damage could impair the normal functions of the DNA and may even result in triggering a program of cell death (apoptosis). The children with this disease do not repair the active genes where oxidative damage occurs. Normally, oxidative damage repair is faster in the active genes (which make up less than five percent of the genome) than in inactive regions of the DNA. The resulting accumulation of oxidative damage could impair the normal functions of the DNA and may even result in triggering a program of cell death (apoptosis).
Genetics
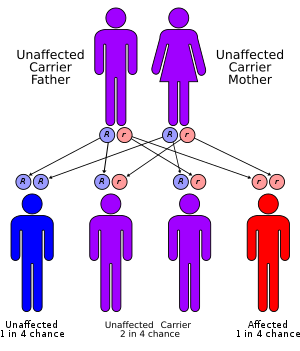
Cockayne syndrome is classified genetically as follows:
| Type | OMIM | Gene |
|---|---|---|
| A | 216400 | ERCC8 (also called CSA) |
| B | 133540 | ERCC6 (also called CSB) |
| C | 216411 | none known |
- Mutations in the ERCC8 (also known as CSA) gene or the ERCC6 (also known as CSB) gene are the cause of Cockayne syndrome.[7] Mutations in the ERCC6 gene mutation makes up ~70% of cases. The proteins made by these genes are involved in repairing damaged DNA via the transcription-coupled repair mechanism, particularly the DNA in active genes. DNA damage is caused by ultraviolet rays from sunlight, radiation, or free radicals in the body. A normal cell can repair DNA damage before it accumulates. If either the ERCC6 or the ERCC8 gene is altered (as in Cockayne Syndrome), DNA damage encountered during transcription isn't repaired, causing RNA polymerase to stall at that location, interfering with gene expression. As the unrepaired DNA damage accumulates, progressively more active gene expression is impeded, leading to malfunctioning cells or cell death, which likely contributes to the signs of Cockayne Syndrome such as premature aging and neuronal hypomyelination.[7]
Mechanism
In contrast to cells with normal repair capability, CSA and CSB deficient cells are unable to preferentially repair cyclobutane pyrimidine dimers induced by the action of ultraviolet (UV) light on the template strand of actively transcribed genes.[11] This deficiency reflects the loss of ability to perform the DNA repair process known as transcription coupled nucleotide excision repair (TC-NER).
Within the damaged cell, the CSA protein normally localizes to sites of DNA damage, particularly inter-strand cross-links, double-strand breaks and some monoadducts.[12] CSB protein is also normally recruited to DNA damaged sites, and its recruitment is most rapid and robust as follows: interstrand crosslinks > double-strand breaks > monoadducts > oxidative damage.[12] CSB protein forms a complex with another DNA repair protein, SNM1A (DCLRE1A), a 5' – 3' exonuclease, that localizes to inter-strand cross-links in a transcription dependent manner.[13] The accumulation of CSB protein at sites of DNA double-strand breaks occurs in a transcription dependent manner and facilitates homologous recombinational repair of the breaks.[14] During the G0/G1 phase of the cell cycle, DNA damage can trigger a CSB-dependent recombinational repair process that uses an RNA (rather than DNA) template.[15]
The premature aging features of CS are likely due, at least in part, to the deficiencies in DNA repair (see DNA damage theory of aging).
Diagnosis
People with this syndrome have smaller than normal head sizes (microcephaly), are of short stature (dwarfism), their eyes appear sunken, and they have a ″aged″ look. They often have long limbs with joint contractures (inability to relax the muscle at a joint), a hunched back (kyphosis), and they may be very thin (cachetic), due to a loss of subcutaneous fat. Their small chin, large ears, and pointy, thin nose often give an aged appearance.[8] The skin of those with Cockayne syndrome is also frequently affected: hyperpigmentation, varicose or spider veins (telangiectasia),[8] and serious sensitivity to sunlight are common, even in individuals without XP-CS. Often patients with Cockayne Syndrome will severely burn or blister with very little heat exposure. The eyes of patients can be affected in various ways and eye abnormalities are common in CS. Cataracts and cloudiness of the cornea (corneal opacity) are common. The loss of and damage to nerves of the optic nerve, causing optic atrophy can occur.[3] Nystagmus, or involuntary eye movement, and pupils that fail to dilate demonstrate a loss of control of voluntary and involuntary muscle movement.[8] A salt and pepper retinal pigmentation is also a typical sign. Diagnosis is determined by a specific test for DNA repair, which measures the recovery of RNA after exposure to UV radiation. Despite being associated with genes involved in nucleotide excision repair (NER), unlike xeroderma pigmentosum, CS is not associated with an increased risk of cancer.[5]
Laboratory Studies
In Cockayne syndrome patients, UV-irradiated cells show decreased DNA and RNA synthesis. https://emedicine.medscape.com/article/1115866-workup#c5 Laboratory studies are mainly useful to eliminate other disorders. For example, skeletal radiography, endocrinologic tests, and chromosomal breakage studies can help in excluding disorders included in the differential diagnosis.
Imaging Studies
Brain CT scanning in Cockayne syndrome patients may reveal calcifications and cortical atrophy.
Other Tests
Prenatal evaluation is possible. Amniotic fluid cell culturing is used to demonstrate that fetal cells are deficient in RNA synthesis after UV irradiation.
Neurology
Imaging studies reveal a widespread absence of the myelin sheaths of the neurons in the white matter of the brain, and general atrophy of the cortex.[5] Calcifications have also been found in the putamen, an area of the forebrain that regulates movements and aids in some forms of learning,[8] along with the cortex.[6] Additionally, atrophy of the central area of the cerebellum found in patients with Cockayne syndrome could also result in the lack of muscle control, particularly involuntary, and poor posture typically seen.
Treatment
There is no permanent cure for this syndrome, although patients can be symptomatically treated. Treatment usually involves physical therapy and minor surgeries to the affected organs, such as cataract removal.[3] Also wearing high-factor sunscreen and protective clothing is recommended because Cockayne Syndrome patients are very sensitive to UV radiation.[16] Optimal nutrition can also help. Genetic counseling for the parents is recommended, as the disorder has a 25% chance of being passed to any future children, and prenatal testing is also a possibility.[3] Another important aspect is the prevention of recurrence of CS in other siblings. Identification of gene defects involved makes it possible to offer genetic counseling and antenatal diagnostic testing to the parents who already have one affected child.[17]
Prognosis
The prognosis for those with Cockayne syndrome is poor, as death typically occurs by the age of 12. [18] The prognosis for Cockayne syndrome varies by the disease type. There are three types of Cockayne syndrome according to the severity and onset of the symptoms. However, the differences between the types are not always clear-cut, and some researchers believe the signs and symptoms reflect a spectrum instead of distinct types: Cockayne syndrome Type A (CSA) is marked by normal development until a child is 1 or 2 years old, at which point growth slows and developmental delays are noticed. Symptoms are not apparent until they are 1 year. Life expectancy for type A is approximately 10 to 20 years. This symptoms are seen in CS type 1 children. Cockayne syndrome type B (CSB), also known as "cerebro-oculo-facio-skeletal (COFS) syndrome" (or "Pena-Shokeir syndrome type B"), is the most severe subtype. Symptoms are present at birth and normal brain development stops after birth. Average lifespan for children with type B is up to 7 years of age. These symptoms are seen in CS type 2 children. Cockayne syndrome type C (CSC) appears later in childhood with milder symptoms than the other types and a slower progression of the disorder. People with this type of Cockayne syndrome live into adulthood, with an average lifespan of 40 to 50 years. These symptoms are seen in CS type 3.
Epidemiology
Cockayne syndrome is rare worldwide. No racial predilection is reported for Cockayne syndrome. No sexual predilection is described for Cockayne syndrome; the male-to-female ratio is equal. Cockayne syndrome I (CS-A) manifests in childhood. Cockayne syndrome II (CS-B) manifests at birth or in infancy, and it has a worse prognosis.
Recent research
The recent research on Jan 2018 mentions different CS features that are seen globally with similarities and differences: CS has an incidence of 1 in 250,000 live births, and a prevalence of 2.5 per million, which is remarkably consistent across various regions globally:[19]
| Affected parts | Clinical features | pathology |
|---|---|---|
| Face | Wizened faceies. Sunken eyes, large ears, thin pointy nose. Small chin. Dental caries, enamel hypoplasia | |
| Skin, hair, nails | Photosensitivity. Wrinkled and aged appearing skin. Thin dry hair, prematurely gray hair. Poor venous access. | |
| Central nervous system | Microcephaly usually beginning at age 2. Mental retardation with low IQ. Delayed milestones.Tremors, ataxia, seizures, strokes, and subdural hemorrhages. | Demyelination – is patchy and segmental– “Metachromatic leukodystrophy". Both oligodendroglia and Schwann cells are affected. Affects cerebral white matter, corpus callosum, brainstem, spinal cord, and peripheral nerves. Neuronal loss at multiple sites, especially the cerebellum. Loss of anterior horn cells due to anterograde and/or retrograde degeneration.
Calcification [55–95%] of the cerebral cortex (especially depths of sulci, basal ganglia, cerebellum, thalamus; also of the arteries, arterioles, and capillaries. Vascular changes - String vessels, especially in areas of Metachromatic leukodystrophy, calcification in leptomeningeal vessels, accelerated atherosclerosis and arteriolosclerosis. Gliosis is present. Astrocytes and microglia may show irregular cytoplasm, multiple nuclei. May be seen as a high-intensity white matter on FLAIR MRI sequences signals. No major brain malformations. Relative sparing of the cerebral cortex, slight thinning of cortical ribbon may be seen. Normal gyral pattern with widening of sulci. Lamination, neuronal size, and configuration of the neocortex are preserved. May show parietal occipital dominance. Severe cerebellar atrophy. Loss of Purkinje, granular neurons, and in some cases neurons in the dentate nucleus. Dendrites of Purkinje cells may be grossly deformed (“cactus flowers”), ferruginated dendrites. Dendrites have fewer higher order branches. Purkinje “axonal torpedoes” may be present. Ventricular enlargement, enlarged cisterna magna are seen. Amyloid plaques, neurofibrillary tangles, Hirano bodies not commonly seen, although ubiquitin reactivity of axons present |
| Hearing and vestibular systems | Sensorineural, high tone hearing loss [60–90%]. Mixed conductive and sensorineural hearing loss (44%) Most commonly bilateral, rarely unilateral | Loss of hair cells in the cochlea, particularly in the basal turn. Loss of neurons in spiral ganglion. Atrophy of auditory pathways. Scala communis, thickened stapes curare, widened prototympanum. Loss of hair cells in pars superior. Loss of neurons in vestibular ganglion. Collapse of the endolymphatic duct of pars inferior |
| Vision | Corneal opacification.
Cataracts [36–86%]. Usually bilateral, most develop by 4 years of age. Pigmentary retinopathy (“salt and pepper”)[43–89%]. Miotic pupils, Optic disk pallor, Enophthalmos, Narrow palpebral fissures. |
Patchy loss of melanin pigment granules. Lipofuscin deposition, large pigment laden cells in a perivascular distribution. Retinal pigment epithelial atrophy and hyperplasia. Loss of cells in ganglion and outer nuclear cell layers. Both the outer and inner segments of photoreceptors are affected. Optic nerve atrophy, with partial demyelination, axonal loss, and gliosis |
| Musculoskeletal system | Cachectic dwarfism. Contractures. Kyphosis, scoliosis. Stooped posture. Muscle wasting. | Denervation myopathy, disuse atrophy |
| Cardiovascular system | Accelerated hypertension. Aortic root dilatation. Cardiomyopathy. | Increased intima medial thickening. Atherosclerosis, arteriosclerosis. |
| Gastrointestinal system | Severe reflux. Abnormal gastrointestinal motility. Many have percutaneous gastrostomy tubes. Hepatomegaly, splenomegaly, elevated liver enzymes. Altered metabolism of drugs | - |
| Renal system | Renal failure | Renal arteries show changes in advanced atherosclerosis and arteriolosclerosis. Unilateral or hypoplastic kidneys. |
| Reproductive system | - | - |
| Males | Micropenis, smaller testicular size | - |
| Females | Ovarian atrophy. A successful pregnancy has been reported. | - |
| Endocrine systems | Normal secondary sexual characteristics. Normal growth hormone, thyroid-stimulating hormone, calcium levels | Normal pituitary gland and thyroid gland |
| Eccrine systems | Decreased production of sweat, tears, saliva | - |
See also
- Accelerated aging disease
- Biogerontology
- Degenerative disease
- Genetic disorder
- CAMFAK syndrome — thought to be a form (or subset) of Cockayne syndrome[20]
References
- Bertola; Cao, H; Albano, Lm; Oliveira, Dp; Kok, F; Marques-Dias, Mj; Kim, Ca; Hegele, Ra (2006). "Cockayne syndrome type A: novel mutations in eight typical patients". Journal of Human Genetics. 51 (8): 701–5. doi:10.1007/s10038-006-0011-7. PMID 16865293.
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews' Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. p. 575. ISBN 978-0-7216-2921-6.
- Bender M, Potocki L, Metry D. What syndrome is this? Cockayne syndrome. Pediatric Dermatology [serial online]. November 2003;20(6):538-540. Available from: MEDLINE with Full Text, Ipswich, MA. Accessed April 30, 2015.
- Hoeijmakers JH (October 2009). "DNA damage, aging, and cancer". N. Engl. J. Med. 361 (15): 1475–85. doi:10.1056/NEJMra0804615. PMID 19812404.
- Nance M, Berry S (1 January 1992). "Cockayne syndrome: review of 140 cases". American Journal of Medical Genetics. 42 (1): 68–84. doi:10.1002/ajmg.1320420115. PMID 1308368.
- Neill CA, Dingwall MM. A Syndrome Resembling Progeria: A Review of Two Cases. Archives of Disease in Childhood. 1950;25(123):213-223.
- Cockayne Syndrome. Genetics Home Reference http://ghr.nlm.nih.gov/condition/cockayne-syndrome Published April 28, 2015. Reviewed May 2010. Accessed April 30, 2015.
- Javadzadeh M. Cockayne Syndrome. Iran J Child Neurol. Autumn 2014;8;4(Suppl.1):18-19.
- Cerebrooculofacioskeletal Syndrome 2. Online Mendelian Inheritance in Man. https://omim.org/entry/610756. Published 2/12/2007.
- Laugel V. Cockayne Syndrome. 2000 Dec 28 [Updated 2012 Jun 14]. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2015. Available from:
- van Hoffen A, Natarajan AT, Mayne LV, van Zeeland AA, Mullenders LH, Venema J (1993). "Deficient repair of the transcribed strand of active genes in Cockayne's syndrome cells". Nucleic Acids Res. 21 (25): 5890–5. doi:10.1093/nar/21.25.5890. PMC 310470. PMID 8290349.
- Iyama T, Wilson DM (2016). "Elements That Regulate the DNA Damage Response of Proteins Defective in Cockayne Syndrome". J. Mol. Biol. 428 (1): 62–78. doi:10.1016/j.jmb.2015.11.020. PMC 4738086. PMID 26616585.
- Iyama T, Lee SY, Berquist BR, Gileadi O, Bohr VA, Seidman MM, McHugh PJ, Wilson DM (2015). "CSB interacts with SNM1A and promotes DNA interstrand crosslink processing". Nucleic Acids Res. 43 (1): 247–58. doi:10.1093/nar/gku1279. PMC 4288174. PMID 25505141.
- Batenburg NL, Thompson EL, Hendrickson EA, Zhu XD (2015). "Cockayne syndrome group B protein regulates DNA double-strand break repair and checkpoint activation". EMBO J. 34 (10): 1399–416. doi:10.15252/embj.201490041. PMC 4491999. PMID 25820262.
- Wei L, Nakajima S, Böhm S, Bernstein KA, Shen Z, Tsang M, Levine AS, Lan L (2015). "DNA damage during the G0/G1 phase triggers RNA-templated, Cockayne syndrome B-dependent homologous recombination". Proc. Natl. Acad. Sci. U.S.A. 112 (27): E3495–504. Bibcode:2015PNAS..112E3495W. doi:10.1073/pnas.1507105112. PMC 4500203. PMID 26100862.
- Kyllermen, Marten. Cockayne Syndrome. Swedish Information Centre for Rare Diseases. 2012: 4.0. http://www.socialstyrelsen.se/rarediseases/cockaynesyndrome#anchor_17 Archived 2015-09-24 at the Wayback Machine
- Title: Cockayne Syndrome Authors: Dr Nita R Sutay, Dr Md Ashfaque Tinmaswala, Dr Manjiri Karlekar, Dr Swati Jhahttp://jmscr.igmpublication.org/v3-i7/35%20jmscr.pdf
- "Cockayne syndrome | Genetic and Rare Diseases Information Center (GARD) – an NCATS Program".
- Karikkineth, A. C.; Scheibye-Knudsen, M.; Fivenson, E.; Croteau, D. L.; Bohr, V. A. (2016). "Cockayne syndrome: Clinical features, model systems and pathways". Ageing Research Reviews. 33: 3–17. doi:10.1016/j.arr.2016.08.002. PMC 5195851. PMID 27507608.
- "Orphanet: CAMFAK syndrome".
External links
- This article incorporates some public domain text from The U.S. National Library of Medicine
| Classification | |
|---|---|
| External resources |