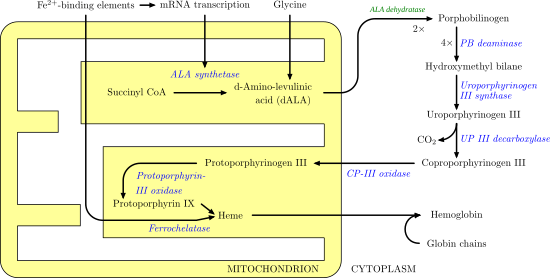
Porphobilinogen deaminase
Porphobilinogen deaminase (hydroxymethylbilane synthase, or uroporphyrinogen I synthase) is an enzyme (EC 2.5.1.61) that in humans is encoded by the HMBS gene. Porphobilinogen deaminase is involved in the third step of the heme biosynthetic pathway. It catalyzes the head to tail condensation of four porphobilinogen molecules into the linear hydroxymethylbilane while releasing four ammonia molecules:
- 4 porphobilinogen + H2O hydroxymethylbilane + 4 NH3

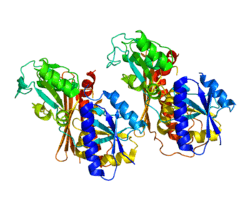


Structure and function
Functionally, porphobilinogen deaminase catalyzes the loss of ammonia from the porphobilinogen monomer (deamination) and its subsequent polymerization to a linear tetrapyrrole, which is released as hydroxymethylbilane:

The structure of 40-42 kDa porphobilinogen deaminase, which is highly conserved amongst organisms, consists of three domains.[5][6] Domains 1 and 2 are structurally very similar: each consisting of five beta-sheets and three alpha helices in humans.[7] Domain 3 is positioned between the other two and has a flattened beta-sheet geometry. A dipyrrole, a cofactor of this enzyme consisting of two condensed porphobilinogen molecules, is covalently attached to domain 3 and extends into the active site, the cleft between domains 1 and 2.[8] Several positively charged arginine residues, positioned to face the active site from domains 1 and 2, have been shown to stabilize the carboxylate functionalities on the incoming porphobilinogen as well as the growing pyrrole chain. These structural features presumably favor the formation of the final hydroxymethylbilane product.[9] Porphobilinogen deaminase usually exists in dimer units in the cytoplasm of the cell.
Reaction mechanism
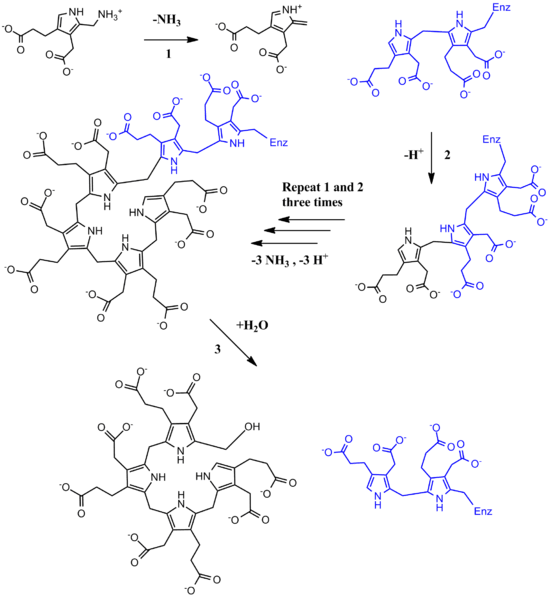
The first step is believed to involve an E1 elimination of ammonia from porphobilinogen, generating a carbocation intermediate (1).[10] This intermediate is then attacked by the dipyrrole cofactor of porphobilinogen deaminase, which after losing a proton yields a trimer covalently bound to the enzyme (2). This intermediate is then open to further reaction with porphobilinogen (1 and 2 repeated three more times). Once a hexamer is formed, hydrolysis allows hydroxymethylbilane to be released, as well as cofactor regeneration (3).[11][12]
Pathology
The most well-known health issue involving porphobilinogen deaminase is acute intermittent porphyria, an autosomal dominant genetic disorder where insufficient hydroxymethylbilane is produced, leading to a build-up of porphobilinogen in the cytoplasm. This is caused by a gene mutation that, in 90% of cases, causes decreased amounts of enzyme. However, mutations where less-active enzymes and/or different isoforms have been described.[13][14][15]
References
- GRCh38: Ensembl release 89: ENSG00000256269 - Ensembl, May 2017
- GRCm38: Ensembl release 89: ENSMUSG00000032126 - Ensembl, May 2017
- "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- Lannfelt L, Wetterberg L, Lilius L, Thunell S, Jörnvall H, Pavlu B, Wielburski A, Gellerfors P (November 1989). "Porphobilinogen deaminase in human erythrocytes: purification of two forms with apparent molecular weights of 40 kDa and 42 kDa". Scand. J. Clin. Lab. Invest. 49 (7): 677–84. doi:10.3109/00365518909091544. PMID 2609111.
- Louie GV, Brownlie PD, Lambert R, Cooper JB, Blundell TL, Wood SP, Warren MJ, Woodcock SC, Jordan PM (September 1992). "Structure of porphobilinogen deaminase reveals a flexible multidomain polymerase with a single catalytic site". Nature. 359 (6390): 33–9. Bibcode:1992Natur.359...33L. doi:10.1038/359033a0. PMID 1522882. S2CID 4264432.
- Gill R, Kolstoe SE, Mohammed F, Al D-Bass A, Mosely JE, Sarwar M, Cooper JB, Wood SP, Shoolingin-Jordan PM (May 2009). "Structure of human porphobilinogen deaminase at 2.8 Å: the molecular basis of acute intermittent porphyria" (PDF). Biochem. J. 420 (1): 17–25. doi:10.1042/BJ20082077. PMID 19207107.
- Jordan PM, Warren MJ (December 1987). "Evidence for a dipyrromethane cofactor at the catalytic site of E. coli porphobilinogen deaminase". FEBS Lett. 225 (1–2): 87–92. doi:10.1016/0014-5793(87)81136-5. PMID 3079571.
- Lander M, Pitt AR, Alefounder PR, Bardy D, Abell C, Battersby AR (April 1991). "Studies on the mechanism of hydroxymethylbilane synthase concerning the role of arginine residues in substrate binding". Biochem. J. 275 (2): 447–52. doi:10.1042/bj2750447. PMC 1150073. PMID 2025226.
- Pichon C, Clemens KR, Jacobson AR, Ian Scott A (June 1992). "On the mechanism of porphobilinogen deaminase. Design, synthesis, and enzymatic reactions of novel porphobilinogen analogs". Tetrahedron. 48 (23): 4687–4712. doi:10.1016/S0040-4020(01)81567-2.
- Battersby AR (December 2000). "Tetrapyrroles: the pigments of life". Nat Prod Rep. 17 (6): 507–26. doi:10.1039/b002635m. PMID 11152419.
- Leeper FJ (April 1989). "The biosynthesis of porphyrins, chlorophylls, and vitamin B12". Nat Prod Rep. 6 (2): 171–203. doi:10.1039/NP9890600171. PMID 2664584.
- "Entrez Gene: HMBS hydroxymethylbilane synthase".
- Grandchamp B, Picat C, de Rooij F, Beaumont C, Wilson P, Deybach JC, Nordmann Y (August 1989). "A point mutation G----A in exon 12 of the porphobilinogen deaminase gene results in exon skipping and is responsible for acute intermittent porphyria". Nucleic Acids Res. 17 (16): 6637–49. doi:10.1093/nar/17.16.6637. PMC 318356. PMID 2789372.
- Astrin KH, Desnick RJ (1994). "Molecular basis of acute intermittent porphyria: mutations and polymorphisms in the human hydroxymethylbilane synthase gene". Hum. Mutat. 4 (4): 243–52. doi:10.1002/humu.1380040403. PMID 7866402.
Further reading
- Deybach JC, Puy H (1995). "Porphobilinogen deaminase gene structure and molecular defects". J. Bioenerg. Biomembr. 27 (2): 197–205. doi:10.1007/BF02110034. PMID 7592566. S2CID 41764609.
- Astrin KH, Desnick RJ (1995). "Molecular basis of acute intermittent porphyria: mutations and polymorphisms in the human hydroxymethylbilane synthase gene". Hum. Mutat. 4 (4): 243–52. doi:10.1002/humu.1380040403. PMID 7866402.
- Helliwell JR, Nieh YP, Habash J, et al. (2003). "Time-resolved and static-ensemble structural chemistry of hydroxymethylbilane synthase". Faraday Discussions. 122: 131–44, discussion 171–90. Bibcode:2003FaDi..122..131H. doi:10.1039/b201331b. PMID 12555854.
- Hessels J, Voortman G, van der Wagen A, et al. (2004). "Homozygous acute intermittent porphyria in a 7-year-old boy with massive excretions of porphyrins and porphyrin precursors". J. Inherit. Metab. Dis. 27 (1): 19–27. doi:10.1023/B:BOLI.0000016613.75677.05. PMID 14970743. S2CID 9504522.
- Kauppinen R (2004). "Molecular diagnostics of acute intermittent porphyria". Expert Rev. Mol. Diagn. 4 (2): 243–9. doi:10.1586/14737159.4.2.243. PMID 14995910. S2CID 21392441.
- Hrdinka M, Puy H, Martasek P (2007). "May 2006 update in porphobilinogen deaminase gene polymorphisms and mutations causing acute intermittent porphyria: comparison with the situation in Slavic population". Physiological Research. 55 Suppl 2: S119–36. PMID 17298216.
- Kauppinen R, Peltonen L, Pihlaja H, Mustajoki P (1993). "CRIM-positive mutations of acute intermittent porphyria in Finland". Hum. Mutat. 1 (5): 392–6. doi:10.1002/humu.1380010508. PMID 1301948.
- Mgone CS, Lanyon WG, Moore MR, Connor JM (1992). "Detection of seven point mutations in the porphobilinogen deaminase gene in patients with acute intermittent porphyria, by direct sequencing of in vitro amplified cDNA". Hum. Genet. 90 (1–2): 12–6. doi:10.1007/BF00210738. PMID 1427766. S2CID 19680295.
- Gu XF, de Rooij F, Voortman G, et al. (1992). "High frequency of mutations in exon 10 of the porphobilinogen deaminase gene in patients with a CRIM-positive subtype of acute intermittent porphyria". Am. J. Hum. Genet. 51 (3): 660–5. PMC 1682727. PMID 1496994.
- Delfau MH, Picat C, De Rooij F, et al. (1991). "Molecular heterogeneity of acute intermittent porphyria: identification of four additional mutations resulting in the CRIM-negative subtype of the disease". Am. J. Hum. Genet. 49 (2): 421–8. PMC 1683312. PMID 1714233.
- Namba H, Narahara K, Tsuji K, et al. (1991). "Assignment of human porphobilinogen deaminase to 11q24.1----q24.2 by in situ hybridization and gene dosage studies". Cytogenet. Cell Genet. 57 (2–3): 105–8. doi:10.1159/000133123. PMID 1914516.
- Lee JS, Anvret M (1992). "Identification of the most common mutation within the porphobilinogen deaminase gene in Swedish patients with acute intermittent porphyria". Proc. Natl. Acad. Sci. U.S.A. 88 (23): 10912–5. doi:10.1073/pnas.88.23.10912. PMC 53042. PMID 1961762.
- Tunnacliffe A, McGuire RS (1991). "A physical linkage group in human chromosome band 11q23 covering a region implicated in leukocyte neoplasia". Genomics. 8 (3): 447–53. doi:10.1016/0888-7543(90)90030-X. PMID 1981047.
- Scobie GA, Llewellyn DH, Urquhart AJ, et al. (1990). "Acute intermittent porphyria caused by a C----T mutation that produces a stop codon in the porphobilinogen deaminase gene". Hum. Genet. 85 (6): 631–4. doi:10.1007/BF00193588. PMID 2227955. S2CID 28858687.
- Delfau MH, Picat C, de Rooij FW, et al. (1990). "Two different point G to A mutations in exon 10 of the porphobilinogen deaminase gene are responsible for acute intermittent porphyria". J. Clin. Invest. 86 (5): 1511–6. doi:10.1172/JCI114869. PMC 296897. PMID 2243128.
- Raich N, Romeo PH, Dubart A, et al. (1986). "Molecular cloning and complete primary sequence of human erythrocyte porphobilinogen deaminase". Nucleic Acids Res. 14 (15): 5955–68. doi:10.1093/nar/14.15.5955. PMC 311614. PMID 2875434.
- Vidaud M, Gattoni R, Stevenin J, et al. (1989). "A 5' splice-region G----C mutation in exon 1 of the human beta-globin gene inhibits pre-mRNA splicing: a mechanism for beta+-thalassemia". Proc. Natl. Acad. Sci. U.S.A. 86 (3): 1041–5. Bibcode:1989PNAS...86.1041V. doi:10.1073/pnas.86.3.1041. PMC 286617. PMID 2915972.
External links