Marginal zone B-cell lymphoma
Marginal zone B-cell lymphomas, also known as marginal zone lymphomas (MZLs), are a heterogeneous group of lymphomas that derive from the malignant transformation of marginal zone B-cells.[1] Marginal zone B cells are innate lymphoid cells that normally function by rapidly mounting IgM antibody immune responses to antigens such as those presented by infectious agents and damaged tissues.[2] They are lymphocytes of the B-cell line that originate and mature in secondary lymphoid follicles and then move to the marginal zones of mucosa-associated lymphoid tissue (i.e. MALT), the spleen, or lymph nodes. Mucosa-associated lymphoid tissue is a diffuse system of small concentrations of lymphoid tissue found in various submucosal membrane sites of the body such as the gastrointestinal tract, mouth, nasal cavity, pharynx, thyroid gland, breast, lung, salivary glands, eye, skin and the human spleen.[3]
| Marginal zone B-cell lymphomas | |
|---|---|
| Other names | Marginal zone lymphomas (MZLs) |
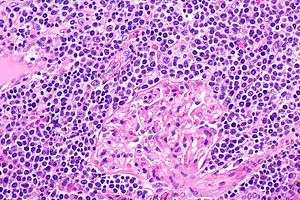 | |
| EMZL infiltrating kidney tissue. | |
| Types | Extranodal marginal zone lymphoma, splenic marginal zone lymphoma, and nodal marginal zone lymphoma. |
In 2016, the World Health Organization classified MZLs into three different types. Extranodal marginal zone lymphomas (EMZLs) are MZLs that develop in extranodal tissues. Most EMZLs develop in MALT and are often termed extranodal MZL of mucosa-associated lymphoid tissue or, more simply, MALT lymphomas. Splenic marginal zone lymphomas (SMZLs) are MZLs that initially are confined to the spleen, bone marrow, and blood.[1] Nodal marginal zone lymphomas (NNMZs) are MZLs initially confined to lymph nodes, bone marrow, and blood.[1] While all of these MZL involve malignant B-cells, they differ not only in the tissues they involve but also in their pathophysiology, clinical presentations, prognoses, and treatments.[1][4]
MZLs represent 5-17% of all Non-Hodgkin lymphomas with the extranodal, splenic, and nodal forms accounting for 50-70%, ~20%, and ~10% of all MZLs.[5] The three MZL subtypes occur more often in older people (age 65–68 years) and are indolent diseases that may in people without symptoms asymptomatic be initially treated by a watchful waiting strategy. However, NMZL carries a somewhat worse long term outcome than the other subtypes[1] and any of the MZL subtypes may progress in a low percentage of cases to a more aggressive lymphoma, particularly diffuse large B-cell lymphoma.[6] One of the most distinctive feature of MLZ is that many cases are associated with the persistent simulation of the immune system by the chronic inflammation that accompanies infections[7] or autoimmune diseases.[8] MZL cases associated with certain infectious pathogens can be cured by treatment directed at the pathogens causing or associated with these infections.[7]
Extranodal marginal zone lymphoma
EMZLs are a form of MZL in which malignant marginal zone B-cells initially infiltrate MALT tissues of the stomach (50-70% of all EMZL) or, less frequently, the esophagus, small intestine, large intestine, rectum, conjunctiva of the eye, nasal passages, pharynx, lung bronchi, vulva, vagina, skin, breast, thymus gland, meninges (i.e. membranes) that envelop the brain and spinal cord, or other organs.[7][9] These EMZLs are classified into subtypes based on the organ(s) involved. For example, EMZL of the stomach is termed primary gastric EMZL. Regardless of subtype, these EMZLs share similar pathophysiological (i.e. disordered physiological processes that cause the disease) and histopathological (i.e. microscopic features of diseased tissues). However, the subtypes differ in presentation, progression, severity, treatment, and instigating factors. The following two sections describe the common pathophysiologic and histopathologic features found in all EMZL subtypes. Features specific to each EMZL subtype follow these two sections.
Pathophysiology
Numerous factors appear involved in the development of EMZL. In a small number of cases, the disease has an increased frequency in patients who have a family history of a hematological malignancy, particularly leukemia, or various autoimmune diseases such as Sjögren syndrome and lupus erythematosus. Inherited genetic, shared environmental, and other as yet unidentified factors may underlie these increased risks of developing an EMZL.[10] Another key factor in the initiation of many EMZL cases is chronic inflammation caused by a chronic infection or autoimmune reaction. The chronic inflammation stimulates B-cells to rearrange their immunoglobulin heavy chain locus so that they encode B-cell receptors that recognize unnatural antigens presented by the injured issues and/or infectious agents that underlie the inflammation. This rearrangement results in the B-cells responding to the ab\normal antigens by taking on features of marginal B-cells and proliferating excessively.[11] In consequence, these B cells progressively acquire in a step-wise fashion chromosome abnormalities, gene mutations, and/or dis-regulated genes that contribute to their becoming malignant. The acquired genomic abnormalities found in EMZL along with the frequency of occurrence in specific EMZL subtypes include the following.
- Chromosome translocations: 1) A translocation of the long (or "q") arm of chromosome 11 at position 21 with the q arm of chromosome 18 at position 21 (notated as a t(11;18)(q21;q21) translocation) occurs in 24% of gastric, 38% of lung, and rarely other EMZL subtypes. This translocation places a part of the API2 gene with a part of the MALT1 gene to create a fusion gene that encodes an Api2-Malt1 fusion protein. This chimeric protein promotes the continuous activation of a transcription factor, NF-κB. NF-κB controls the expression of various genes which increase the survival, cytokine production, and other potentially malignant behaviors of cells. 2) A t(14;18)(q32;q21) translocation occurs in 7% of ocular adnexa, 6% of lung, and rarely if at all in other cases of EMZL. It causes the overexpression of Malt1. This protein indirectly inhibits programmed cell death to prolong cell survival and also promotes activation of NF-κB. 3) A t(1;14)(p22;q32) ("p" stands for a chromosome's short arm) translocation occurs in ∼9% of lung, ∼4% of stomach, and rarely if at all in other cases of EMZL. This translocation causes the overexpression of the BCL10 gene. Bcl10 protein contributes to the activation of NF-κB. 4) A t(3;14)(p13;q32) translocation occurs in rare cases of EMZL and is thought to cause overexpression of the FOXP1 gene. FoxP1 protein stimulates production of transcription factors such as PRDM1, IRF4, and XBP1 which promote the maturation of B-cells to plasma cells. 5) Three translocations, t(1;14)(p21;q32), t(5;14)(q34;q32), t(9;14)(p24;q32), and t(X;14)(p11.4;q32), occur in rare cases of EMZL but their effects on promoting malignancy are unknown.[12]
- Gene inactivations and mutations: 1) Inactivation or the TNFAIP3 gene due to its deletion on chromosome 6 about position 23 (i.e. a 6q23 deletion) or its mutation occur mainly in ocular adnexa, salivary gland, and thyroid gland EMZL. TNFAIP3 inactivation generally takes place in cases which do not have any of the above chromosome translocations. This gene's product, tumor necrosis factor, alpha-induced protein 3, functions to impair the activation of NF-κB. 2) Gain-of-function mutations in the MYD88 gene occur in ~5% of ocular adnexa EMZL cases. The product of this gene, myeloid differentiation primary response 88, continuously activates NF-κB as well as the STAT3 and AP1 transcription factors.[12] 3) Inactivating mutations in the NOTCH1 (8% of all cases) and NOTCH2 (8% of all cases) genes occur in EMZL. The products of these genes are cell surface receptor proteins which when bound to their activating ligands move to the cell nucleus and contribute to the activation of genes that control the development, proliferation, survival, and migration of B cells.[13]
Many EMZL subtypes are associated with infectious agents or autoimmune diseases that may contribute to their malignant development. The following Table reports on these EMZL subtypes; the tissues they involve; the infectious agents/autoimmune diseases that may underlie the development of the EMZL subtypes; the strength of evidence linking these infectious agents/autoimmune diseases to their malignancy; the incidence (i.e. percentage) of cases with the EMZL subtype associated with the infectious agent/autoimmune disease; and some of the chimeric genes expressed by the neoplastic B-cells of the EMZL subtype.
| Subtype | Tissue(s) involved | Infectious agent or autoimmune diseasen[7] | Strength of evidence | Incidence[7][14] | Chimeric genes expressed (percentage of cases)[14] | |
|---|---|---|---|---|---|---|
| Primary gastric EMZL | stomach | Helicobacter pylori | confirmed[7][15] | ~80% | BIRC3-MALT1 (23%), IGH-FOXP1 (3%), IGH-BCL-10 (2%), and IGH-MALT1 (1%) | |
| Primary gastric EMZL | stomach | Helicobacter heilmannii sensu lato | likely[7][14] | <1% | similar to the antibiotics used for Helicobacter pylori[16] | |
| Primary salivary gland EMZL | salivary and lacrimal glands | Sjögren syndrome | confirmed[7][17] | ~4.3% | IGH-MALT1 (6%), BIRC3-MALT1 (2%), and IGH-BCL-10 (1%) | |
| Primary thyroid EMZL | thyroid gland | Hashimoto's thyroiditis | confirmed[7][17] | ~0.5% | ICH-FOXP1 (50%) and BIRC3-MALLT1 (9%) | |
| Primary ocular adnexa EMZL | ocular adenexa (i.e. orbit, conjunctiva, and eyelids) | Chlamydia psittaci | suggestive[7][18] | 10-50% | IGH-FOXP1 (20%), IGH-MALT1 16%), and BIRC3-MALT1 (7%) | |
| Primary cutaneous EMZL | skin | Borrelia burgdorferi | suggestive[18] | variable | IGH-FOXP1 (10%), IGH-MALT1 (7%), and BIRC3-MALT1 (4%) | |
| Primary small intestinal EMZL | small intestine | Campylobacter jejuni | low[18][3] | variable | BIRC3-MALT1 (19%) and IGH-BCL10 (7%) | |
| Primary pulmonary EMZL | lung | Achromobacter xylosoxidans | low[19][20] | <50% | BIRC3-MALT1 (45%), IGH-BCL10 (8%), and IGH-MALT1 (7%) | |
Histopathology
The histopathologic (i.e. microscopic) examination of EMZL lesions typically reveals a vaguely nodular or diffuse pattern of cells. The malignant cells in these lesions have, in varying proportions, the morphology of small-to-medium-sized lymphocytes, centrocyte-like B cells, centroblast-like B cells, monocyte-like B cells, plasma cell-like B cells, and/or large B cells. When the large B cells form prominent sheets that are clearly separated from cells with the other, low-grade malignant morphologies, the disease may be transforming to the far more aggressive malignancy, diffuse large B-cell lymphoma. This transformation occurs in ~18% of patients at a median of 4–5 years after the original diagnosis of EMZL.[21] Immunophenotyping or the neoplastic large B cells in these lesions shows that they express CD20 but not CD3 surface membrane B cell marker proteins. The cells almost always express BCL2 and may express MNDA (~70% of cases), CD23 (~33% of cases) and CD5 (~20% of cases) marker proteins but do not express the cyclin D1 marker protein.,[21] the T-cell marker, CD10, or BCL6.[1]
Subtypes, diagnosis, treatment, and prognosis
There are various EMZL subtypes based on the organs they involve. Almost all of these subtypes occur in the mucosa-associated lymphoid tissue of the affected organ and are often termed MALT lymphomas of the affected organ (e.g. gastric MALT lymphoma). However, these lymphomas are also termed primary EMZL of the affected organ (e.g. primary gastric EMZL). While both terms are used here for these subtypes, the primary (organ involved) EMZL is preferred to indicate that the EMZL subtype initially developed in and may remain limited to the indicated tissue. However, approximately 30% of cases disseminate to other sites, predominantly lymph nodes and in rare cases the bone marrow. The malignant B-cells in these subtypes may also circulate in the blood but this is very uncommon. Regardless of the subtype of EMZL or its dissemination to other tissues, the prognosis for these lymphomas is good with 5 year overall survivals generally ranging between 86% and 95%.[14][6]
Primary gastric

Primary gastric EMZL, also termed primary gastric MALT lymphoma or, more often, just gastric MALT lymphoma, is usually an indolent disease that in ~10% of cases also involves other GI tract and/or non-GI tract sites. Patients commonly present at an early stage of the disease[22] with various symptoms such as nausea, vomiting, indigestion, upper abdominal pain, and gastric bleeding as indicated by coughing up blood, bloody bowel movements, and/or iron deficiency anemia. Rarely, patients present with perforation of the stomach or B symptoms such as fever and night sweats. Individuals with chronic Helicobacter pylori infection may also have halitosis.[23] Endoscopic inspection and biopsy of lesions[15] and endoscopic-based ultrasound[22] inspection of the upper GI tract show lesions, most often in the stomach's pyloric antrium, that are superficial mucosal erosions, shallow ulcers, nodules, enlarged rugae, and/or thickenings of the stomach wall.[15] The histopathology of primary gastric MALT lesions and the marker proteins and genomic abnormalities expressed by the malignant cells in these lesions are given in the Histopathology section. Primary gastric EMZL is associated with infection of the stomach with Helicobacter pylori in >80% of cases or with Helicobacter heilmannii sensu lato in <1% of cases.[14] Indications that gastric Helicobactor pylori is the causes for gastric EMZL include: a positive urea breath test; a positive stool test that detects an antigen of the pathogen in the patient's feces; a positive urease test in a biopsied tissue specimen; a positive serum or whole blood test using specific antibodies directed against the pathogen; and growth of the pathogen in tissue cultures of a biopsied tissue.[24] Helicobacter heilmannii sensu lato designates at least 11 different Helicobactor species of which 5 are known to infect the human stomach. It has been more difficult to determine that Helicobacter heilmannii sensu lato is responsible for human gastric disease because the urea breath test is less often positive in infestations by these species, antibodies directed against them are generally not available, and they are difficult to grow in culture. The diagnosis of Helicobacter heilmannii sensu lato therefore depends upon detecting the organism in tissues or fecal material histologically using special silver staining methods and then sequencing certain genes (i.e. urease A, urease B, heat shock protein 60, and/or gyrase subunit B) in the organisms DNA and/or the organisms 23sRNA.[16] The 'Treatment of localized (i.e. Ann Arbor stage I and II) Helicobactor pylori-positive primary gastric EMZL employs any one of several different Helicobacter pylori eradication protocols. These protocols include a proton-pump inhibitor (e.g. omeprazole or lansoprazole[25]) plus any one of several different antibiotic combinations (e.g. Clarithromycin + Amoxicillin or levofloxacin + nitazoxanide + doxycyclin).[15] The exact drug regimen is chosen based on the known or suspected resistance of the pathogen to these antibiotics in individual cases. The regimen is given for 7–14 days and followed-up by testing within 4 weeks for the presence of the pathogen by the urea breath or stool antigen test. If the initial regimen fails to eradicate the pathogen, patients are treated with a second regimen using a combination of three or four drugs (e.g. a proton-pump inhitor + bismuth subcitrate + tetracycline + metronidazole).[15] Eradication of the pathogen is successful in 70-95% of cases. Recently, a sequential treatment regimen (i.e. a proton-pump inhibitor + amoxicillin followed by a proton-pump inhibitor + clarithromycin + Tinidazole) has been reported to eradicate the pathogen in >90% of cases.[15] Patients who have lesions that harbor a t(11;18) or t(1;14) chromosomal translocation and therefore express the BIRC3-MALT1 or IGH-BCL10 chimeric protein, respectively, have an increased incidence of being resistant to Helicobactor pylori eradication protocols.[14] Some 50-80% of patients who have experienced eradication of the pathogen develop within 3–28 months a remission and long-term clinical control of their lymphoma. Radiation therapy to the stomach and surrounding (i.e. peri-gastric) lymph nodes has been used to successfully treat: a) localized Helicobactor pylori-positive primary gastric EZML in which the pathogen has not been eradicated by the cited drug protocols; b) localized Helicobactor pylori-negative primary gastric EZML cases; and c) Helicobactor pylori-positive and -negative EZML cases in elderly or frail patients. Patients with systemic (i.e. Ann Arbor stage III and IV) primary gastric EMZL who are free of symptoms have been treated with watchful waiting or if symptomatic been treated with the immunotherapy drug, rituximab (given for 4 weeks) combined with chlorambucil given for 6–12 months; 58% of these patients attain a 58% progression-free survival rate at 5 years. Frail stage III/IV patients have been successfully treated with rituximab or cyclophosphamide alone.[1] While the treatment of Helicobactor heilmanni sensu lato depends on far fewer studies, it generally follows the recommendations used for the treatment of Helicobactor pylori.[16]
Primary small intestinal
Primary small intestinal MZL, also termed primary small intestinal MALT lymphoma, commonly presents with colicky abdominal pain, diarrhea, and in cases of advanced disease signs and symptoms of malabsorption (e.g. weight loss, malnutrition, and anemia), small bowel obstruction, ascites (i.e. fluid in the abdominal cavity), and/or enlargements of lymph nodes, spleen, and/or liver.[26] While generally a progressive disease, patients with early stage primary small intestinal MZL may have spontaneous and complete remissions.[7] Immunoproliferative small intestinal disease, formerly termed Mediterranean lymphoma or considered a type of alpha heavy chain disease (IgA/αHCD),[27] is a variant and by far the most common form of small intestinal MZL.[26] This variant is endemic in countries of the Mediterranean Basin, particularly those of the Middle East although cases of this disease have been found throughout the world usually but not always in immigrants from the Middle East.[26] In its endemic areas, immunoproliferative small intestinal disease constitutes ~30% of all GI tact lymphomas, mainly afflicts individuals 20–30 years old who are of low socioeconomic status, and is associated with infection by the food-borne bacterium, Campylobacter jejuni. Campylobacter jejuni-associated disease is more prevalent in individuals who express human leukocyte antigen AI19, B12, or A9 or are blood type B. It is suggested that these individuals are genetically predisposed to developing the disease.[18] Immunostaining of small bowel lesions in these cases commonly detects the presence of Campylobacter jejuni and is predictive that the disease will respond to antibiotics. However, it is not clear that this bacterium is the actual cause of immunoproliferative small intestinal disease:[26] it may merely colonizes the gut of individuals with the disease while other as yet unidentified antibiotic-sensitive bacteria or non-bacterial pathogens, e.g. parasites, underlie the disease's development.[26]
In primary small intestinal EMZL cases, double-balloon enteroscopy and capsule endoscopy reveal the presence of extensive mucosal erosions and/or, less commonly, polyps, nodules, masses, and/or scarring.[22] These lesions localizes to the duodenum, jejunum, or ilium in about 63, 17, and 8% of cases, respectively, or involve more than one small intestinal site in ~17% of cases.[26] The lesions consist of lymphocytes, atypical plasma cells and, less commonly, centrocyte-like cells infiltrates in the intestinal lamina propria[18] with the lymphocytes and centrocyte-like cells expressing marker proteins (e.g. CD20 and CD79a) that are typical for EMZL.[26] Campylobacter jejuni is detected in these lesions by immunostaining. Patients with this disease commonly have a monoclonal gammopathy as evidence by the presence of a monoclonal antibody consisting of the fragment crystallizable region of the IgA heavy chain in their blood, jejunum juice, and/or, rarely, urine. The abnormal IgA protein is detected in patients' sera by immunofixation using an antibody directed against the heavy chain fragment of IgA.[26]
The Treatment of primary small intestinal EMZL has focused on nutritional support and control of symptoms including surgery and/or radiotherapy to treat bowel obstructions and highly localized disease. However, studies indicate that individuals with the disease, particularly those with the immunoproliferative small intestine disease form, have overall response rates of ~90% following treatment with broad-spectrum antibiotics such as tetracycline, metronidazole, or tetracycline + ampicillin.[3] These responses are durable in most cases. Accordingly, antibiotic therapy is recommended to treat early stage disease. Patients refractory to antibiotic therapy have been treated with chemotherapy (i.e. CHOP or a CHOP-like regimes) followed by long-term maintenance with tetracycline. This treatment regimen has achieved 5 year overall survival rates of 70%. Since surgery and radiotherapy are not curative for the disease, high dose chemotherapy regimens and autologous stem cell transplantation have been recommended for refractory and/or relapsed disease.[2]
Colorectal EMZLs
ENZLs involving the colon or rectum are extremely rare. In a 2019 review of 73 cases, persons diagnosed with one of these ENZL subtypes had a median age of 62 years (range 26-72), were predominately female (66%), and had their primary tumors located in the rectum (74% of cases), right colon (13.6%), transverse colon (4.1%), or sigmoid colon (8.2%). Thirty percent of these individuals had multiply tumors, ~40% of which were in sites in the gastrointestinal tract outside of the colon and rectum. These individuals were initially treated and achieved complete remissions with local surgical resection in 18 of 19 cases, more extensive surgical resection in 18 of 19 cases, chemotherapy in 12 of 13 cases, radiation therapy (in 4 of 5 cases, or antibiotic therapy to achieve Helicobacter pylori eradication in 12 of 15 cases. Two individuals received no treatment with one of these having a spontaneous remission. The 8 cases not achieving a complete remission required second-line treatment; 3 cases were remission failures.[28] The following sections further describe these two EMZL subtypes.
Primary colonic
Primary colonic EMZL, also termed primary colonic MALT lymphoma, usually presents at an early stage of disease with evidence of lower GI tract bleeding (e.g. tarry bowel movements and/or iron deficiency anemia), less commonly with lower abdominal pain, and rarely with bowel perforation or intussusception. Endoscopic examination most often reveals a single polyp or rarely multiple polyps, a mucosal ulcer, or a mucosal nodule. Diagnosis is passed on biopsy of the lesions showing a histology typical of EMZL, e.g. diffuse infiltrates composed of small to medium-sized lymphocytes that may show morphological features of monocytes and/or plasma cells. The lymphocytes in these lesions express B cell markers (e.g. CD19 and CD79a) typical of EMZL lesions. The best treatment regimen for this lymphoma is debated. Surgical resection, endoscopic resection, radiation, and chemotherapy have been employed. Surgery followed by chemotherapy (mitoxantrone + chlorambucil + prednisone or cyclophosphamide + vincristine + prednisone combined with either chlorambucil or rituximab) have been regarded as first-line treatment for the disease. More recently, rituximab alone as a single agent has also been found successful in treating primary colonic MALT lymphoma. Finally, rare cases of primary colonic EMZL have been completely resolved using Helicobacter pylori antibiotic therapy.[9]
Primary rectal
Primary rectal EMZL, more commonly termed MALT lymphoma of the rectum, usually presents at an early stage of disease with anal bleeding and/or blood in the stool. Endoscopic examination reveals a rectal polyp, rectal mass, or, less commonly, a rectal ulcer.[9] Some cases of this lymphoma have been reported to regress spontaneously.[29] On examination, >90% of cases present with localized (i.e. stage I or II) disease. The lymphomas' lesions are characterized by reactive lymphoid follicles infiltrated with centrocyte-like or monocyte-like B cells (the latter cells may show features of plasma cells). The malignant cells in these lesions may contain the t(11;18) translocation and therefore express the API2-MALT1 chimeric protein (11% of cases). Some 22-45% of casests are associated with Helicobactor pylori GI tract infection.[30] Treatments for the disease have included radiotherapy, surgical resection, endoscopic mucosal resection, various chemotherapies, and antibiotic-based eradication of Helicobactor pylori. Eradication therapy for Helicobactor pylori-positive cases using currently recommended standard antibiotic regimens has given complete responses in 12 of 19 cases and therefore is regarded as proper treatment for such cases. Surgical resection for localized disease has achieved long-term survivals in individual cases.[30] However, radiotherapy for localized disease has given 5-year disease-free and overall survival rates of 76% and 96%, respectively, in 16 of 19 reviewed cases[30] and is suggested to be the preferred treatment for patients suffering Helicobactor pylori-positive primary rectal EMZL.[9]
Primary esophagus
Primary EMZL of the esophagus, also termed MALT lymphoma of the esophagus, is extremely rare with most cases of it being reported from Japan. It presents with symptoms of difficul swallowing and/or sensations of a foreign body in the esophageal area. Endoscopy, endosonography and chest CT scans reveal a solitary esophageal mass of varying size[31] or, more commonly, a linear central indentation or ridge in the esophagus.[32] In a 2017 review, 6 of 18 patients with EMZL of the esophagus had evidence of concurrent Helicobacter pylori infection. The histopathology of the lesions in EMZL of the esophagus is typical of EMZL in showing the presence of centrocyte-like cells, monocyte-like cells, and small lymphocytes that express CD20 but not CD10.[31] Treatment of EMZL of the esophagus has consisted of endoscopic resection, surgical resection, radiotherapy, endoscopic resection plus radiotherapy, or chemotherapy. Most patients show a complete response to these interventions. However, the long-term efficacy of these responses is not known since treatment follow-up times have been short (6–35 months). Systematic antibiotic-based eradication therapy to treat Helicobactor pylori-associated EMZL of the esophagus had not been reported[31] until a recent case with the disease was treated with vonoprazan + amoxicillin + clarithromycin for 1 week. The patient showed evidence of eradicating the bacterium based on a urea breath test but nonetheless subsequently evidenced progression the lymphoma.[33]
Primary ocular adnexa
Primary ocular adnexa EZML (also termed primary EMZL of the ocular adnexa, primary ocular adnexa MALT lymphoma, or primary MALT lymphoma of the ocular adnexa) occurs primarily in older patients (median age 65 years). Individuals may be predisposed to the disease by having a long history of exposure to livestock, mainly cattle and pigs or working with meat from these animals; autoimmune diseases, particularly autoimmune thyroid disease; and infections, particularly Clamydophelia psittaci, a communicable intracellular bacterium that infects feral birds, farm animals, and humans. In humans, it causes respiratory psittacosis and eye infections, particularly chronic conjunctivitis.[34] Clammydophelia psittaci has been detected in the lesions of 47-80% of patients with primary ocular adnexa EMZL with the highest rates of this detection occurring in Italy, Austria, Germany, and Korea. Much lower detection rates are reported in the United Kingdom and Southern China while there has been little or no evidence of this organism in cases from the United States and Japan.[14] Gastric Helicobactor pylori infection or Hepatitis C virus infection have been reported to be associated with primary ocular adnexa EZZML in ~33% and 2-36%, respectively, of cases; in rare cases, the disease has also been associated with Herpes simplex virus 1, Herpes simplex 2, adenovirus 8, adenovirus 19, Chlamydia trachomatis, or Chlamydophila abortus infection. The relationship(s) of these infections to the development and/or progression of primary ocular adnexa EZML is unclear.[34]
Patients present with conjunctival (25% of cases) or intra-orbital (75% of cases) lesions which typically involve one eye but in 10-15% of cases, particularly in conjunctival cases, involve both eyes. Conjunctiva lesions usually present as a salmon red patch that covers the outer layer of the eyeball; intra-orbital lesions commonly present as exophthalmos (i.e. a bulging of the eye) (27% of cases), a palpable eye mass (19%), ptosis (i.e. eyelid drooping) (6%) and/or, less commonly, diplopia (i.e. double vision), impaired orbital mobility, excessive tearing, and/or orbital nodules.[34] Patients with conjunctiva disease may be asymptomatic.[35] A variable percentage of patients with primary ocular adnexa MZL may concurrently be afflicted with Hashimoto's thyroiditis, Sjögren syndrome, or IgG4-related disease. Some studies have also found that the disease is associated with Helicobactor pylori infection of the stomach (45% of cases) or EMZL in other tissues (25% of cases). The lesions in primary ocular adnexa EMZL are typical of EMZL: they contain centrocyte-like B-cells, monocyte-like B-cells, and/or small lymphocytes many of which express CD20, CD791, PAZ5, and BCL2 but not CD10 or cyclin D1 proteins.[36] The t(1:14)(p22:q320 chromosome translocation, which leads to the over expression of the MALT1 gene, trisomy 3, trisomy 18, and deletions at position 23 on the long arm of chromosome 6 are also often found in primary ocular adnexa EMZL.[34]
Treatment of the disease requires further study. In patients with localized disease: 1) radiotherapy has achieved complete responses in 52-93% of cases and 5 year systemic-free (but not local-free) relapse rates of >90%; 2) chemotherapy with a CHOP regimen in 15 patients has achieved remissions without relapse in 9, local relapse in 5, and systemic relapse in 2 patients after 55 months of follow-up; 3) chemotherapy with chlorambucil in 33 patients has achieved complete responses in 26 patients after 24 months of follow-up; 4) immunotherapy with rituximab has achieved variable results over the short-term and requires further study over longer follow-up periods[36] with somewhat better results occurring in patients with conjunctival disease;[34] and 5) antibiotic therapy with doxycycline has achieved 2 year and 5 year failure-free survival rates of 67% and 55%, respectively, and a 5-year progression-free rate of 61%.[36] The generally recognized treatment of choice for patients with systemic involvement uses various chemotherapy regimens often combined with rituximab. Complete responses have been observed in most patients treated with chlorambucil, CHOP regimens, or rituximab but recurrence rates have been high (e.g. ~33%).[34]
Primary skin
Primary cutaneous EMZL or primary cutaneous MALT lymphoma (also termed skin-associated lymphoid tissue lymphoma) typically presents as single or multiple small papules or plaques usually located on the arms and/or trunk. Histologically, these lesions consist of reactive germinal centers containing a mixture of small B cells that have a plasma cell-like or monocyte-like morphology interlaced with numerous T cell lymphocytes.[37] The B-cells in these lesions express the B-cell markers commonly seen in EMZL.[38] The DNA of Borrelia burgdorferi, the causative agent of Lyme disease, has been detected in the lesions of 10%–42% of patients[39] in Germany, Italy, Japan, and Turkey but not of patients from Spain, Finland, the Netherlands, or the United States.[40] While the disease almost always has a highly indolent course, it is subject to repeated relapses that are usually limited to the skin. Rarely, primary cutaneous EMZL disseminates to other tissues and becomes a systemic disease.[40] Treatment of primary cutaneous EMZL has been conservative, given the diseases indolent behavior. In Borrelia burdorferi-positive disease, antibiotic therapy (courses of a cephalosporin or tetracycline are regarded as first-line antibiotic choices) should be considered[7] although this measure is less applicable in areas, such as the United States, where the bacterium has not been associated with the disease.[38] Treatment of Borrelia burdorferi-negative disease or disease in which where antibiotic therapy is not an option or has failed depend on the extent of the lesions. Single lesion treatments include surgical resection, direct injection of interferon-alpha or rituximab into the lesions, and localized external beam radiotherapy. Disease-free rates found 5 and 10 years after these treatments are 57% and 43%, respectively. In disseminated disease, watchful waiting, intravenous rituximab, and chemotherapy have been used. Intravenous rituximab achieves remission rates of 85% in patients that have not received chemotherapy; chlorambucil chemotherapy with or without intravenous rituximab has significantly improved event-free survival times; and CHOP chemotherapy is recommended for patients with B symptoms (e.g. fever, night sweats, weight loss, etc.), elevated levels of serum lactic acid dehydrogenase, or disease that has progressed to a more aggressive stage.[7]
Primary lung
Primary pulmonary EMZL (or primary pulmonary MALT lymphoma) is a rare disorder but nonetheless represents up to 80% of all lymphomas originating in the lung. The cause for developing this lymphoma is unclear. Some 16% of individuals with the disease present with features of an autoimmune disorder with one study reporting that 57 of 124 patients with the disorder evidenced Achromobacter xylosoxidans DNA in their lung lesions.[41] Achromobacter xylosoxidans is a betaproteobacteria that is routinely isolated from the lungs of cystic fibrosis patients; it has a low virulence but is extremely resistant to antibiotics.[7] Primary pulmonary EMZL usually afflicts patients 50–60 years old; in almost 50% of cases the disease is diagnosed in symptom-free individuals who present with an abnormal chest X-ray or CT scan conducted for unrelated reasons.[41] Patients with symptoms usually present with chest pain, shortness of breath, and/or a history of recurrent respiratory infections.[14] Chest X-rays and CT scans typically show bilateral alveolar opacities that are <5 cm. More severe lung damage (e.g. atelectasis, pleural effusion, or mediastinal lymphadenopathy) occur in <10% of cases. Patients with more advanced disease may present with bone marrow involvement (13-30% of cases), involvement or other sites outside the lung such as the stomach, eyes, ears, nose, and/or throat (25-35% of cases), or in rare cases of particularly aggressive disease, systemic B symptoms such as fever, night sweats, and/or weight loss. A monoclonal gammopathy (i.e. excessive amounts of a monoclonal gamma globulin in the blood) is found in 20-60% of cases, occurring particularly in individuals with tissue lesions that contain lymphocytes that have a plasma cell-like appearance. The lesions in primary pulmonary lymphoma are in the mucosa of the broncheal airways and are diagnosed by needle biopsy, bronchial biopsy, trans-bronchial biopsy, and/or bronchoalveolar lavage. Findings consistent with the diagnosis include biopsy specimens revealing mucosa infiltrates of small B-cells bearing the typical B cell markers found in EMZL; occasional specimens consist of B lymphocytes with a plasma cell appearance. Bronchoalveolar lavage fluid may contain >10% of cells which bear these markers. B-cells in the pulmonary lesions have the t(11;18)(q21;q21) translocation and therefore express the API2-MALT1 chimeric protein in ~40% of cases. Other, less frequently occurring genomic abnormalities in these cells include t(1;14)(p22;q32), t(14;18)(q32;q21), and t(3;14)(p14.1;q32) translocations and trisomy of chromosomes 3 and/or 18.[41]
Treatment of primary pulmonary EMZL varies. Antibiotic therapy has not been studied and cannot be recommended. Recommended treatments which have afforded overall 5 year survival rates of 89-100%[42] include surgery, radiotherapy, chemotherapy, immunotherapy, and watchful waiting.[41] Surgical resection or radiotherapy may be considered for localized disease. In more extensive disease, rituximab immunotherapy has achieved a 70% response rate but with a high rate of recurrence (~36%). Treatment with fludarabine, CHOP, chlorambucil, or chlorambucil + rituximab have been used to treat extensive disease with each treatment giving approximately similar overall median survival times of >10 years.[41]
Primary salivary gland
Primary salivary gland EMZL (also termed MALT lymphoma of the salivary glands) or, in cases primarily involving the lacrimal glands, primary lacrimal gland EMZL (also termed MALT lymphoma of the lacrimal gland) is a complication of Sjögren syndrome, an autoimmune disease characterized by chronic inflammation of the salivary and/or lacrimal glands.[17] This autoimmune disease is thought to be caused by a combination of genetic and environmental factors including infectious agents. This lymphoma afflicts ~3% of patients with Sjögren syndrome[8] and involves one or more of the afflicted salivary or, less commonly, lacrimal glands.[17] The median time between diagnosis of the autoimmune disease and EMZL varies between 7.5[17] and 11 years.[19] Afflicted individuals typically are 55–60 years old and present with localized hardening and/or enlargement of the parotid gland or, less commonly, other salivary or lacrimal gland. Some 20% of cases present with or progress to involve local lymph nodes or the spleen to cause lymphadenopathy or splenomegaly while ~10% of cases present with or progress to a high-grade lymphoma, primarily diffuse large B-cell lymphoma.[17] Histologically, the involved glands show lymphocyte-based lesions that are typical of EMZL with the infiltrating lymphocytes in some cases having morphological features resembling plasma cells. In individuals with more advanced disease, these lesions develop in the mucosal linings of the eye socket, nasal cavity, pharynx, airways of the lower respiratory tract, stomach, and/or thyroid gland.[8]
Treatment of primary salivary/lacrimal gland EMZL has not been standardized. A minority of patients have been treated by watchful waiting but most patients have been subjected to surgery, radiotherapy, chemotherapy (i.e. chlorambucil), immunotherapy (i.e. rituximab, or a combination (e.g. chlorambucil + rituximab or fludarabine + rituximab or bendamustine + rituximab) immunotherapy plus chemotherapy regimen. In general, overall survival rates after 5, 10, and 15 years of treatment have been 95%, 85%, and 78% respectively. While the response to these therapeutic regimens has been very good, ~33% of treated patients have experience a recurrence of their lymphoma in the salivary/lacrimal glands, lymph nodes, or other sites.[19]
Lymphoepithelial sialadenitis
Lymphoepithelial sialadenitis, also termed chronic sialadenitis, is a benign infiltration of salivary glands by B-cells with morphological features of marginal zone B-cells, centrocytes, and monocytes. Histologically, this disorder is associated with atrophy of the columnar epithelium in salivary gland ducts as well as the proliferation of epithelial cells and lymphoepithelial lesions in these glands. While usually a component of Sjorgen syndrome, these histological finding can also occur in patients without evidence of this syndrome. Very rarely, lymphoepitheleial sialadenitis progressed to salivary gland EMZL.[14]
Primary thyroid
Primary thyroid EMZL, also termed MALT lymphoma of the thyroid gland, is extremely rare. It occurs almost exclusively in thyroid glands afflicted by Hashimoto's thyroiditis, an autoimmune disease characterized by the accumulation of lymphocytes, including B-cells, in the thyroid gland and the subsequent destruction of thyroid tissue by these cells.[43] Patients with this syndorme have a 40- to 80-fold increased risk of developing a thyroid lymphoma, 25% of which are primary thyroid EMZLs. Hashimoto's thyroiditis patients who develop this lymphoma are typically women (median age 70 years) who have had the thyroiditis for 20–30 years and present with a rapid increase in the thyroid gland's size and, in association with this, have developed hoarseness, high-pitched breath sounds, and/or difficulty in swallowing and/or breathing.[17] Histologically, the lesions in this lymphoma generally consist of reactive lymphoid follicles and lymphoepithelial lesions which are populated by intermediate-sized B-cells, centrocytes, plasma cells, and, in ~1/3 of cases, sheets of large lymphocytic cells similar to those seen in diffuse large B-cell lymphomas. The malignant cells in these lesions express B-cell markers that are typical for EMZL, e.g. CD20 and BCL-6 but not CD10 proteins.[44] Patients with primary thyroid EMZL are at an increased risk of developing a more disseminated lymphoma, particularly diffuse large B-cell lymphoma or, alternatively, nodal MZL, or splenic MZL.[17]
Treatment of primary thyroid EMZL is generally conservative since up to 90% of patients are diagnosed with early stage disease.[17] Although the optimal treatment for this disease is uncertain, the majority of patients with localized disease are treated with surgery, radiotherapy, or a combination of both modalities and attain overall response rates of up to 100%[19] and an estimated 5 year disease-free survival of 95%.[17] Surgery plus radiotherapy does not appear to give better results that radiotherapy alone.[44] Patients with extensive disease or disease that has progressed to a higher grade lymphoma (principally diffuse B-cell lymphoma) have been treated with chemotherapy (usually CHOP[44][45]) and/or immunotherapy (i.e. rituximab[44]). However, the 5 year survival rates in chemotherapy-treated patients with disseminated primary thyroid EMZL[46] or disease that has progressed to a more malignant lymphoma[17] are only 35% and 44%, respectively.
EMZL and other autoimmune diseases
Patients suffering an autoimmune disease other then Hashimoto's thyroiditis also have an increased risk of developing an EZML in one or more tissue sites. For example, patients with systemic lupus erythematosus have a 7.5-fold increased risk of developing an EMZL compared to the general population and at diagnosis of this development have a median age of 50 years and been diagnosed with systemic lupus erthyematosis for 6.7 to 17.8 years.[17] Patients with rheumatoid arthritis,[17] immune thrombocytopenic purpura,[47] and autoimmune hemolytic anemia[48] are similarly susceptible to developing an EZML. While the exact reasons for these associations are unclear, it is generally considered that the chronic inflammation involved in each disease promotes the malignant behavior of B-cells and thereby the development of EZML.[17] Treatment of patients with an autoimmune disease complicated by EMZL has usually involved the standard measures used to treat both the autoimmune disease and EMZL.[48]
Primary central nervous system
Primary EMZL of the central nervous system is an extremely rare disorder. Compared to other central nervous system lymphomas which are highly aggressive, primary EMZL of the central nervous system is a non-aggressive, low-grade lymphoma. In a review of 70 published cases, the disease involved the proliferation of malignant marginal zone B-cells within the dura mater, i.e. thick membrane surrounding the brain and spinal cord, (56 cases), the brain or spinal parenchyma (6 cases), the brain's cavernous sinusn (4 cases), the brains choroid plexus (3 cases), inside brain [[Ventricular system[ventricle]] (1 case), the cerebellopontine angle (2 cases), and the optic nerve (2 cases). Patients (77% female; median age 55 years, ranging from 18 to 78 years) presented with various neurological signs and symptoms depending on the site of involvement. The most common presenting symptoms were headache (30 cases); seizures (22 cases); and visual changes (19 cases). Less commonly, patients presented with paresthesias (i.e. abnormal skin sensations), motor deficits, and ataxias, memory failures, and dizziness. At the time of diagnosis, there was no evidence of EMZL outside of the central nervous system. Malignant cells were detected in the cerebrospinal fluid in 5 of the 19 cases tested for this.[49] Histologically, lesions in the disorder were typical of EMZL in that they consisted of small to medium-sized B-cells that express CD19, CD20, and CD79a) but not CD10, CD23, or cyclin D1 marker proteins along with some plasma cells and a variable number of reactive T-cells.[49][50] Fifty percent of cases tested for trisomy of chromosome 3 were positive.[49]
Treatment of localized disease consisted of surgery, radiotherapy, or a combination of both modalities whereas treatment of extensive central nervous system disease consisted of chemotherapy, including intrathecal chemotherapy, with or without surgery and/or radiotherapy. Regardless of treatment regimen, primary central nervous system EZML has a good prognosis with complete response (CR) occurring in 77% of patients and 22% of patients alive with evidence of disease after 1–86 months of follow-up times. The values of systemic and intrathecal chemotherapy in treating the disease are unclear and require further study.[49]
Primary breast
Primary EMZL of the breast (also termed primary MALT lymphoma of the breast) is an exceedingly rare disease. It usually presents as a palpable breast mass in an otherwise symptom-free patient.[51] Histopathological findings are typical for EMZL: lesions consist of small- to medium-sized B cells, centrocyte-like B-cells, small lymphoid cells with some features of plasma cells or monocytes, and mature plasma cells with the lymphoid cells in these lesions expressing CD20 and CD79a but usually not CD10, CD43 or BCL6 marker proteins.[52] Moderate doses of local radiation therapy are recommended to treated localized EMZL of the breast. This treatment has achieved overall survival rates of >90%. Given these results and the high sensitivity of EMZL to radiation therapy, mastectomy is not recommended and wide excision is not usually necessary to treat localized disease. For patients with disseminated disease, treatment options include watchful waiting and chemotherapy (typically employing a CHOP or CHOP-like regimen) with or without radiation therapy and/or excision. These approaches have attained complete disease remissions in 9 of 9 patients followed for 6–74 months and one death due to progressive disease in a patient followed for 107 months. Other drugs used to treat the disease include rituximab, tamoxifen, and oxaliplatin.[51]
Primary urinary tract
Primary urinary tract EMZLs of the urinary bladder and kidney are extremely rare but the most common forms of lymphoma that are found in these organs. They occur most commonly in middle aged females who have a history of chronic cystitis, i.e. inflammation of the bladder due to urinary tract infection or other causes.[19]
Primary bladder
Presenting symptoms of primary bladder lymphoma include weight loss, fatigue, hematuria, dysuria, nocturia, urinary frequency, and pain in the abdomen and/or suprapubic area.[53] However, this lymphoma commonly occurs as a disseminated disease involving other organs and tissues.[54] Radiological and cystoscopy examinations reveal one or more mucosal masses in, or diffuse thickening of, the bladder wall.[19] The histopathology of these lesions is typical of EMZL; they contain small lymphocytes some or many of which have plasma cell features with the malignant cells in these lesions typically expressing CD20 and PAX-5 but not CD5 or CD10 marker proteins.[53] The cells may also contain the t(11;18)(q21:q 21) translocation typical of EZML.[54] Treatment of primary bladder EMZL depends on the extent of disease. Localized disease should be confirmed using, e.g. Positron emission tomography–computed tomography (i.e. PET/CT), Magnetic resonance imaging (i.e. MRI) of the pelvis area, and Bone marrow examination. Confirmed localized disease has been treated by surgery and radiotherapy with radiotherapy being the clearly preferred and most appropriate modality given this lymphoma's high sensitivity to radiation. However, surgical resection with resection of bladder tumor (i.e. TURBT) may be the best treatment where fertility is of concern. Disseminated and recurrent primary bladder EMZL have been treated with systemic chemotherapy (usually a CHOP or CHOP + rituximab regimen.[53] Prognoses for treated localized and disseminated disease are good[19] with long-term (e.g. up to 40 years) remissions reported for most patients with localized disease and (up to 10 years) for patients with disseminated disease.[53]
Kidney
Kidney EMZL (i.e. kidney MALT lymphoma, renal EMZL, or renal MALT lymphoma) occurs primarily in individuals >50 years old but has been reported in individuals as young as 9 years. In slightly more than half of the reported cases, this lymphoma was localized to the kidney or detected in the kidney plus lymph nodes around the kidney, elsewhere in the retroperitonium, or along the abdominal aorta. These cases could therefore be regarded as primary kidney EMZLs. The remaining cases had widespread disease some of which appear related to primary salivary gland EMZL, primary orbital EMZL, Helicobactor pylori-associated gastritis, systemic lupus erythematosus, or possibly an Epstein–Barr virus-associated lymphoproliferative disease, i.e. a lymphocyte-proliferating disease related to and thought to be caused by infection with this virus.[55] Patients may present with signs and symptoms of a kidney mass (e.g. low pack pain and/or abnormal kidney function as determined by elevation in serum creatinine). The best treatment for kidney EMZL is unclear. Reported cases have been subjected to nephrectomy and/or chemotherapy.[56]
Primary gallbladder
Primary gallbladder EMZL (i.e. extranodal marginal zone lymphoma of the gallbladder,[57] primary MALT lymphoma of the gall bladder[58]) is an extremely rare disease with only 17 cases being reported in the literature as of 2017.[58] Presenting features in individuals (aged 31–84 years, median age 74, >60% females) with the disease are similar to those seen in other lymphomas and non-lymphomatous cancers of the gallbladder;[57] these include pain in the upper right-side of the abdomen, nausea, vomiting, and in about two-thirds of cases, gallstoness.[58] Given these similarities as well as similarities in the laboratory, medical ultrasound, and X-ray findings of primary gallbladder EMZL compared to other gallbladder cancers which account for >99% of bladder cancers, the diagnosis of primary gallbladder EMZL has not yet been made pre-operatively. Rather, its diagnosis has rested exclusively on examination of surgically-removed gallbladders.[58] The lesions in these gallbladders show infiltrates in the gland's submucosa that consist of small lymphocytes interspersed with lymphoepithelial lesions. The lymphocytes have marker protein profiles (e.g. CD20 and Bcl-2 positive; CD5, cyclin D1[58] and CD10[57] negative) that are typical for EMXL. Cholecystectomy, i.e. surgical removal of the gallbladder, has produced remissions in all patients with only one recurrence over observation periods of 2 to 96 months.[58]
Primary hepatic
Primary hepatic EMZL (i.e. primary hepatic extranodal marginal zone B-cell lymphoma, primary hepatic mucosa-associated lymphoid tissue lymphoma, primary hepatic mucosa-associated lymphoma) is an extremely rare malignancy representing <3% of all primary lymphomas of the liver.[59] Only 47 cases of primary hepatic EMZL were reported in the English literature as evaluated by a 2019 review.[60] Based on this review, individuals with primary hepatic EMZL had concomitant liver disease ( principally hepatitis B viral hepatitis or hepatitis C viral hepatitis, less commonly, primary biliary cirrhosis or hepatocellular carcinoma, and, rarely, other liver diseases[60] such as hepatitis A viral hepatitis. Patients (median age 63 years, range 30–85 years) presented with no symptoms (~64% of cases) or symptoms (which may have been related to their other liver diseases) such as abdominal pain, generalized weakness, cough, elevations in their blood levels of liver enzymes, and/or evidence of one or more liver masses as detected by magnetic resonance imaging, computed tomography scans, or positron emission tomography.[60] These presentations are virtually identical to those seen in other forms of liver cancer. Accordingly, the diagnosis of primary hepatic EMZL has been extremely hard to make without obtaining tissue by surgical methods.[59] Histological examination of involved liver tissues commonly showed diffuse infiltrations of small- to medium-sized atypical lymphocytes. These infiltrations, which may involve the liver's bile ducts, often contained lymphoepithelial lesions. Immunohistochemistry testing of these tissues revealed lymphocytes that expressed CD20 and BCL-2 but not CD10 or cyclin D1. While optimal therapeutic strategies for this disease have not been established, primary hepatic EMZL appears to be an indolent cancer. Patients who underwent surgical resection with or without chemotherapy or rituximab treatment regimens and were observed over a median period of 31 months had mostly positive outcomes: 92% survived, 8% died of causes unrelated or only indirectly related to their cancer, and 11% suffered relapses.[60]
EMZL associated with hepatitis C
EMZL occurs more frequently (~2.5-fold increased risk) in individuals who have hepatitis C virus-induced hepatitis. The lymphoma typically occurs 15–25 years (median times) after the viral infection and involves the skin (35% of cases), salivary glands (25%), orbital adnexa (15%) or, uncommonly, the stomach or other tissues. It is associated with type II cryoglobulinemia, i.e. the circulation of an immune complex consisting of polyclonal IgG, monoclonal IgM, and hepatitis C viral RNA. This immune complex causes signs and symptoms of vasculitis in 10% of cases. Other signs and symptoms of the disorder include those associated with chronic hepatitis and the specific subtype of EMZL.[61] Rarely EMZL associated with hepatitis C virus infection presents as single or multiple soft, mobile sub-cutaneous nodules.[27] This presentation occurs mainly in female (83% of cases) and elderly patients.[61] Patients with this disorder may have detectable levels of circulating hepatitis C virus.[39] The histology of the lesions in EMZL associated with hepatitis C virus infection is typical of EMZL[39] although the genomic abnormalities in the disorders malignant cells has not been well-defined beyond their expression of the t(14;18) chromosome translocation in a significant number of cases.[62] Treatment of this disease had relied on eradicating the virus using peginterferon-alfas, interferon-alpha-like drugs to mobilize the hosts' immune systems. This treatment cured the viral infection in ~50% and produced lymphoma remissions in <50% of cases. More recently, drugs (e.g. simeprevir, daclatasvir, sofosbuvir, and dasabuvir) that directly inhibit the virus's reproduction have cured the infection and achieved lymphoma responses in up to 100 and 73%, respectively, of patients with one year overall and progression-free survival rates of 98 and 75%, respectively. For patients who's lymphoma fails to respond to this therapy (~25% of cases), recommended treatments include rituximab or rituximab + a peginterferon-alfa. Since chemotherapy regimens are highly toxic in patients with liver disease, they should be avoided, where possible, in treating EMZL associated with hepatitis C virus infection.[61]
Splenic marginal zone lymphoma
Splenic marginal zone lymphoma (SMZL) is a low grade lymphoma in which malignant B-cells accumulate in the spleen, bone marrow, and, less commonly, the circulation. While generally an indolent disease, about 5-10% of cases transform into a far more aggressive malignancy, diffuse large B-cell lymphoma.[63] In a variable percentage of cases, SMZL has been observed to occur with an increased incidence in individuals who are chronically infected with Hepatitis C virus[62] or have any one of various chronic autoimmune diseases or abnormalities.[64]
Signs and symptoms
At presentation, patients (median age 65 years; range 30–90 years) generally exhibit enlargement of their spleens (75% of cases).[65] They typically do not have enlargements of their lymph nodes except for the lymph nodes around the hilum of their spleens.[66] Most patients have no systemic symptoms such as fever, night sweats, weight loss, or fatigue.[65] Blood tests reveal reductions in the levels of red blood cells, platelets, and/or white blood cells in 25% of cases;[65] an abnormal circulating IgM myeloma protein in <33% of cases; and in ~20% of cases evidence of autoimmunity abnormalities such as circulating autoantibodies (i.e. antibodies directed against the patients' own antigens), autoimmune hemolytic anemia, immune thrombocytopenic purpura,[65] cold agglutinins, and/or anticoagulant antibodies.[64] Individuals with SMZL also commonly exhibit increased levels of circulating blood lymphocytes which in some cases can be identified as malignant B-cells; these malignant cells may have hair-like projections similar to the malignant B-cells found in the circulation of patients with hairy cell leukemia.[66] Patients with SMZL may also present with signs and symptoms of acquired von Willebrand disease, angioedema due to C1-esterase inhibitor deficiency,[64] or hepatitis C virus infection (e.g. clinical hepatitis, circulating hepatitis C virus). The association of hepatitis C virus infection with SMZL varies with location and may be as high as 10% in some areas.[62] Finally, careful examination of patient bone marrows almost always finds pockets or more extensive accumulations of malignant B-cells.[65]
Pathophysiology
The malignant cells involved in SMZL are tentatively identified as antigen-experienced B-cells. The disease appears to be initiated in at least some cases by chronic antigen stimulation of the precursor B-cells that thereby become antigen-experienced. Evidence for this derivation comes from studies showing that the antigen-experienced B-cells inn SMZL express structurally restricted immunoglobulins and B-cell receptors (see clonal selection) that likely bind specific but generally unidentified antigens.[67] Furthermore, patients with SMZL are often found to have autoimmune abnormalities such as circulating autoantibodies (i.e. antibodies directed against patients' own antigens), autoimmune hemolytic anemia, immune thrombocytopenic purpura,[65] cold agglutinin disease, circulating anticoagulant antibodies, acquired von Willebrand disease, and angeoedema due to C1-esterase inhibitor deficiency.[64] It is thought that the B-cell receptor binding of unidentified antigens including those involved in the cited autoimmune abnormalities stimulate the B-cells' proliferation, long-term survival, and thereby the step-wise acquisition of genomic abnormalities which ultimately cause the antigen-experienced B-cells to become malignant.[63][67][65] The genomic abnormalities thought to contribute to this malignant transformation include:
- Chromosome abnormalities such as: 1) deletions in the long (i.e. "q") arm of chromosome 7 (annotated as del7q) in 30-40% of cases (this deletion is rare in other lymphomas and therefore used as a marker for SMZL);[63] 2) deletion of a region on the short (i.e. "p") arm of chromosome 17 in 3-17% of cases to result in a loose of one of the two p53 genes that encode a tumor suppressor that acts to regulate cellular survival; and 3) gains in the q arm of chromosome 3 in 10-20% of cases.[65]
- Mutations in genes such as: 1 KLF2 (21% of cases),[63] a transcription factor that indirectly regulates the NF-κB signaling pathway of cellular survival, proliferation, and the production of cell-stimulating cytokines;[68] 2) NOTCH2 (20% of cases),[63] a membrane protein that regulates the development of marginal zone B-cells from their precursor cells and has tumor suppressor activity to thereby promote cellular survival;[69] 3) TP53 (14% of cases), a transcription factor that indirectly regulates cell proliferation and programmed cell death to thereby promote cellular survival;[63] 4) IGLL5 (14% of cases) with unclear functions;[63] 5) TNFAIP3 (13% of cases), which acts indirectly to inhibit NF-κB activation and programmed cell death;[63] and 6) in <10% of cases at least 16 other genes.[63]
Overall, mutations in the NOTCH, NF-κB, and KLF2 signaling pathways appear particularly important in the pathogenesis of SMZL.[64]
Diagnosis
The clearest evidence for the diagnosis of SMZL is obtained by examination of patients' spleens obtained by splenectomy. These spleens characteristically show lymphoid infiltrates in the white pulp and, to a lesser and more variable extent, the red pulp. These infiltrates consist of small lymphocytes, marginal zone B-cells, centroblast-like B-cells, monocyte-like B-cells, and plasma cells. Epithelioid-like histiocytes may be found in the red pulp. Splenic hilar lymph nodes may show nodular infiltrates of small lymphocytes. Careful and thorough examination of patients' bone marrow commonly shows aggregates of lymphoid cells between the organs trabeculae and within its sinuses. Neoplastic B-cells may also circulate in patient's blood. The neoplastic cells in all of these tissues, similar the neoplastic cells in extranodal and nodal MZL, express CD20, CD27, and BCL2 but not CD10, CD23, CD5, CD43, CD38, BCL6, cyclin D1, or annexin A1 marker proteins. These cells may also express the del7q deletion (i.e. deletions in the q arm of chromosome 7) in 30-40% of cases[65] and in lower percentages of cases the mutated genes listed in the Pathophysiology section. While the diagnosis of SMZL was initially based on the examination of splenic tissue, currently the diagnosis is made in most cases on clinical findings plus examinations of patients' bone marrow and/or blood that detect neoplastic B-cells that express some of the proteins and/or genomic abnormalities cited above;[65] however, cases difficult to diagnose based on bone marrow and blood findings require examination of the spleen to obtain a definitive diagnosis of SMZL.[66]
Treatment
Given its rarity, there have been no systematic and controlled studies on the treatment of SMZL. Current recommendations for this include the following. Watchful waiting, which is the withholding of specific treatments while performing follow-up examinations every 3 to 6 months to detect disease progression. This course is recommended for the ~33% of SMZL patients who present with asymptomatic, non-progressive, or slowly progressive disease. These patients may not require therapeutic interventions for long periods. Historically, the initial therapy for patients with rapidly progressing disease was splenectomy. Some 90% of these patients show reductions in their symptoms and improvements in their low red blood cell, platelet, and white blood cell counts; they had median progression-free, 5 year overall, and 10 year overall survival rates of 8.2 years, 84%, and 67%, respectively. However, these patients show no alteration in the neoplastic B-cells levels in their blood, were subject to serious complications from their splenectomy (e.g. thrombosis, infections), and did not show alter overall survival rates better than those obtained with other treatment strategies. Accordingly, splenectomy for SMZL has been limited to cases of significantly symptomatic enlarged spleens in patients with mild-to-moderate bone marrow involvement and no bulky lymph node enlargements.[66]
Current treatment recommendations for patients with symptomatic or rapidly progressive SMZL rely on drugs. Rituximab, a commercial preparation of a monoclonal antibody directed at the CD20 protein on B-cells, is significantly active in SMZL, with short-term treatments (e.g. ~4 weeks) achieving overall response rates of 90-100%, complete remission rates of >50%, and a 7-year progression-free survival rate of 69%. Long-term maintenance therapy with rituximab appears to improve these results and patients who relapse after rituximab therapy commonly respond to a second course of the drug. Prior to rituximab availability, single drug chemotherapy (e.g. chlorambucil, cyclophosphamide, fludarabine, pentostatin, 2CDA, or bendamustine) and multiple drug regimens (i.e. the CVP regimen of cyclophosphamide, vincristine, and prednisone, or the CHOP regimen of CVP plus doxorubicin) were used to treat the disease. However, current studies indicate that these chemotherapeutic agents are not superior to single agent rituximab therapy in terms of response rate as well as the quality and duration of these responses.[66] A phase II clinical trial has found that the treatment of SMZL with a combination of rituximab plus bemdamustine achieves overall response and complete response rates of 91% and 73%, respectively, with percentage responses enduring for >3 years, progression-free survival rate, and overall survival rate of 93%, 90%, and 96%, respectively. The results of this trial, while requiring confirmation, strongly suggest that this two-drug regimen be used in place of rituximab alone, the cited chemotherapy regimens, or rituximab plus the cited chemotherapy regimens for patients with symptomatic/progressive SMZL.[70]
Experts recommend that SMZL patients who also have hepatitis C virus infection should be treated with drugs that act to eliminate the virus as their first line approach. Before the development of directly acting anti-viral agents, several studies reported that IFN-α treatment of these patients produced improvements not only in the viral infection but also remissions (~65% of cases) in their lymphomas.[66] Several newer, directly acting anti-viral agents, e.g. grazoprevir, daclatasvir, sofosbuvir, and dasabuvir, are more effective in treating hepatitis C viral infection[61] and in s small number of patients have been or are expected to be more effective in producing lymphoma remissions in patients with SMZL plus hepatitis C virus infection.[61][66]
Nodal marginal zone lymphoma
Nodal marginal zone lymphoma (NMZL), previously termed monocytoid B-cell lymphoma, nodal monocytoid B-cell lymphoma, and nodal marginal zone lymphoma with or without monocytoid B-cells, is an infiltration of tissues with malignant lymphoid cells that have the morphological and phenotypical features of all marginal zone lymphomas.[71] NMZL differs from the other subtypes of marginal zone lymphomas by its primary involvement of lymph nodes rather than other tissues and organs.[72] NMZL is the least common subtype of the three marginal zone lymphomas.[73]
Signs and symptoms
Almost all patients with NMZL present (median age 50–64 years;[71] male to female ration 1.5 to 1[73]) with non-bulky enlargement of their lymph nodes in the neck, groin, abdomen, and thoracic regions;[71] some cases may also exhibit this involvement in their Waldeyer's tonsillar ring.[74] Patients at presentation are generally fully functional but in 10-20% of cases complain of B symptoms such as fever, night sweats, weight loss, and/or fatigue.[74] Laboratory studies show malignant B[cells infiltrating the bone marrow in ~33% of patients and an abnormal IgM myeloma protein in ~20% of cases. Rarely, patients may present with circulating malignant marginal zone B cells and/or reductions in one or more types of circulating normal blood cells.[71] Biopsy of involved tissues various patterns (e.g. diffuse throughout lymph node, centered between the follicles of lymph nodes, and/or a nodules spread throughout the lymph node) lymphocyte infiltration.[74] These patterns are similar to those seen in EMZL MALT lymphomas.[73] The cells in these infiltrations ore, in varying proportions, small lymphocytes, marginal zone-like B-cells, centrocyte-like cells, monocyte-like cells, plasma cell-like cells, and in >20% of cases large blastic B-cells. The malignant B-cells in these infiltrations are, like those in other marginal zone lymphomas, Marginal zone B-cells that typically express CD20, CD19, CD79, and Bcl2 but not CD10, CD5, CD23, or cyclin D1.
Pathophysiology
Some 6-19% of NMZL cases have been reported to be associated with autoimmune diseases such as rheumatoid arthritis, Sjögren syndrome, autoimmune hemolytic anemia, and chronic thyroiditis. However, there is little evidence that these diseases contribute to the development of NMZL.[74] Furthermore, the association of NMZL with hepatitis C virus infections found in earlier studies has not been confirmed in more recent studies.[73] It therefore appears that the postulated role of chronic immune stimulation in promoting extranodal and splenic marginal zone lymphomas has not been clearly demonstrated in and may not apply to NMZL: the underlying initiating cause for developing this disease is currently unclear. Nonetheless, instigating B-cells in NMZL acquire genomic abnormalities that are thought to contribute to their malignant transformation. These genomic abnormalities include the following.
- Chromosomal abnormalities such as: 1) trisomy of chromosome 3 (24% of cases) which causes the overexpression of FOXP1, NFKBIZ, and BCL6 whose protein products promote cellular proliferation and survival;[71] 2) trisomy of chromosome 18 (~50% of cases) causing the overexpression of NFATC1[71] whose protein product may act to promote cell proliferation and survival;[75] 3) uncommonly, trisomy of chromosomes 7 and 12 and deletion of the long arm of chromosome 6 which have as yet unknown functional effects;[71] and 4) chromosomal translocation between the short (i.e. "p") arm of chromosome 2 at position 24 and the long (i.e. "q") arm of chromosome 14 at position 32, a translocation of as yet unknown functional consequence but not found in the other marginal zone lymphoma forms and therefore useful as a diagnostic marker for NMZL.[76]
- Mutations in genes such as: 1) NOTCH2 (25% of cases)[63] a membrane protein that regulates the development of marginal zone B-cells from their precursor cells and also is a tumor suppressor which acts to regulate cellar survival;[69] 2) TNFAIP3 (5-15% of cases) whose product is a deubiquitinating enzyme that functions to suppress the NF-κB transcription factor and thereby the NF-κB signaling pathway which controls cellular activation, proliferation, and survival;[67] 3) BIRC3, which encodes the cIAP2 protein that functions to regulate cell death caused by apoptosis;[67] 3) MYD88 (0-10% of cases) whose protein product indirectly regulates activation of the NF-κB cell signaling pathway;[67] 4) KLF2 whose product protein is a transcription factor which indirectly regulates the NF-κB cell signaling pathway;[67] 5) PTPRD whose product protein is a receptor tyrosine phosphatase that has tumor suppressor activity and indirectly regulates several signaling programs that regulate cell proliferation and responses to cytokines;[67] and 5) in ~40% of cases one or more of various other genes such as MLL2, SIN3A, ARID1A, EP300, CREBBP, and TBL1XR1) that have chromatin remodeling activity to thereby regulate the expression of a wide range of other genes.[67]
Diagnosis
The diagnosis of NMZL depends upon identifying neoplastic B-cells in lymph nodes and in some cases the bone marrow but not, at least in early stage disease, in extra-nodal organs. These neoplastic cells should express the marker proteins common to marginal zone lymphomas (refer to previous section) and, in most cases, one or more of the genomic abnormalities indicated in the Pathophysiology section.[71]
Treatment
Recommended treatments for NMZL depend on the diseases state. Asymptomatic NMZL may use watchful waiting with routine follow-up examinations every, e.g. 3–6 months, to check for disease progression. However, localized disease, even in asymptomatic patients, has been successively treated with surgery followed by local radiotherapy. Disease that moves past a localized stage to become disseminated, rapidly progressive disease, and symptomatic disease have been treated with a single chemotherapy drug (e.g. cladribine, fludarabine, chlorambucil, or bendamustine); a single immunotherapy drug (e.g. rituximab); a multiple drug chemotherapy regimen (e.g. CHOP), or a combination multiple chemotherapy drug plus immunotherapy drug regimen (i.e. CHOP + rituximab). It is not clear that any one or more of these regimens achieves is superior to the others.[73]
Prognosis
NMZL is considered an incurable but relatively indolent disease that takes a slowly progressive, relapsing course. Its prognosis appears to be slightly worse than that seen in extranodal and splenic marginal zone lymphomas[71] with ~15% of people progressing to a more aggressive lymphoma, diffuse large B cell lymphoma, at median time of ~4.5 years after the diagnosis of NMZL.[73] In different studies, people with the disease have 5 year survival rates of 62-90%.[73]
Children
In children NMZL has been classified by the World Health Organization (2016) as a separate variant of NMZL based on its presentation, histology of the involved lymph nodes, and clinical course.[73] Of the >60 published cases, 95% of Pediatric NMZL cases occurred in adolescent boys with >90% of cases presenting as an asymptomatic, localized (Stage I/II) disease involving enlargement of the lymph nodes of the head and neck regions. These cases showed no associations with autoimmune or pathogen-induced inflammatory diseases. All of these findings contrast with those seen in extranodal marginal zone lymphomas occurring in children. Histologically, the involved lymph nodes show infiltrations in the germinal centers of afflicted lymph nodes by lymphoid cells usually expressing CD20 and CD43, often (~50% of cases) expressing Bcl2, and usually not expressing CD10 or BCL6. The marginal zone B-cells in these infiltrations have relatively few genomic abnormalities compared to NMZL in adults. Trisomy of chromosome 18 has been reported in 21% of cases and, in rare cases, trisomy of chromosome 3. No recurrent gene mutations have been reported to occur in these cells.[77] The course of pediatric NMZL is extremely indolent with the disease having a low relapse rate and typically an excellent outcome.[73] Observation perionds of up to 12–18 years have found that patients have overall survival rates of 100% and relapse rates of ~4%.[77] Treatment of pediatric NMZL has used a watchful waiting strategy, rituximab, chemotherapy, and/or local radiation therapy. The watchful waiting strategy has done as well as the other therapies and is therefore the recommended initial treatment for the disease.[77]
Recent research
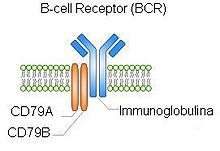
Various new drugs such as B-cell receptor (see adjacent figure) signaling blockers and ibritumomab tiuxetan (Zevlin) are being tested in clinical trials for MZL.[78] These trials are important in determining dosages and safety of the drugs in study. As of January 19, 2017, the FDA approved the first ever targeted drug for MZL, ibrutinib.[79] This drug works by inhibiting Bruton's tyrosine kinase (BKT), which is able to send signals to the nucleus for survival. In other words, it slows the growth of B-cells.[79] Vaccines have been developed that greatly reduce the number of Helicobacter pylori in the stomach of animals previously colonized with these bacteria. One or more of these vaccines may be a promising candidates to control Helicobacter pylori infection in humans as well as farm and domesticated animals.[80]
References
- Bron D, Meuleman N (September 2019). "Marginal zone lymphomas: second most common lymphomas in older patients". Current Opinion in Oncology. 31 (5): 386–393. doi:10.1097/CCO.0000000000000554. PMID 31246587.
- Thieblemont C, Zucca E (2017). "Clinical aspects and therapy of gastrointestinal MALT lymphoma". Best Practice & Research. Clinical Haematology. 30 (1–2): 109–117. doi:10.1016/j.beha.2017.01.002. PMID 28288705.
- Sriskandarajah P, Dearden CE (2017). "Epidemiology and environmental aspects of marginal zone lymphomas". Best Practice & Research. Clinical Haematology. 30 (1–2): 84–91. doi:10.1016/j.beha.2016.07.002. PMID 28288721.
- Swerdlow SH, Campo E, Pileri SA, Harris NL, Stein H, Siebert R, Advani R, Ghielmini M, Salles GA, Zelenetz AD, Jaffe ES (May 2016). "The 2016 revision of the World Health Organization classification of lymphoid neoplasms". Blood. 127 (20): 2375–90. doi:10.1182/blood-2016-01-643569. PMC 4874220. PMID 26980727.
- Cheah CY, Opat S, Trotman J, Marlton P (February 2019). "Front-line management of indolent non-Hodgkin lymphoma in Australia. Part 2: mantle cell lymphoma and marginal zone lymphoma". Internal Medicine Journal. 49 (9): 1070–1080. doi:10.1111/imj.14268. PMID 30816618.
- Casulo C, Friedberg J (2017). "Transformation of marginal zone lymphoma (and association with other lymphomas)". Best Practice & Research. Clinical Haematology. 30 (1–2): 131–138. doi:10.1016/j.beha.2016.08.029. PMID 28288708.
- Smedby KE, Ponzoni M (November 2017). "The aetiology of B-cell lymphoid malignancies with a focus on chronic inflammation and infections". Journal of Internal Medicine. 282 (5): 360–370. doi:10.1111/joim.12684. PMID 28875507.
- Nocturne G, Pontarini E, Bombardieri M, Mariette X (March 2019). "Lymphomas complicating primary Sjögren's syndrome: from autoimmunity to lymphoma". Rheumatology (Oxford, England). doi:10.1093/rheumatology/kez052. PMID 30838413.
- Abbas H, Niazi M, Makker J (May 2017). "Mucosa-Associated Lymphoid Tissue (MALT) Lymphoma of the Colon: A Case Report and a Literature Review". The American Journal of Case Reports. 18: 491–497. doi:10.12659/AJCR.902843. PMC 5424574. PMID 28469125.
- Bracci PM, Benavente Y, Turner JJ, Paltiel O, Slager SL, Vajdic CM, Norman AD, Cerhan JR, Chiu BC, Becker N, Cocco P, Dogan A, Nieters A, Holly EA, Kane EV, Smedby KE, Maynadié M, Spinelli JJ, Roman E, Glimelius B, Wang SS, Sampson JN, Morton LM, de Sanjosé S (August 2014). "Medical history, lifestyle, family history, and occupational risk factors for marginal zone lymphoma: the InterLymph Non-Hodgkin Lymphoma Subtypes Project". Journal of the National Cancer Institute. Monographs. 2014 (48): 52–65. doi:10.1093/jncimonographs/lgu011. PMC 4207869. PMID 25174026.
- Bertoni F, Rossi D, Zucca E (2018). "Recent advances in understanding the biology of marginal zone lymphoma". F1000Research. 7: 406. doi:10.12688/f1000research.13826.1. PMC 5874504. PMID 29657712.
- Du MQ (2017). "MALT lymphoma: Genetic abnormalities, immunological stimulation and molecular mechanism". Best Practice & Research. Clinical Haematology. 30 (1–2): 13–23. doi:10.1016/j.beha.2016.09.002. PMID 28288707.
- Arruga F, Vaisitti T, Deaglio S (2018). "The NOTCH Pathway and Its Mutations in Mature B Cell Malignancies". Frontiers in Oncology. 8: 550. doi:10.3389/fonc.2018.00550. PMC 6275466. PMID 30534535.
- Schreuder MI, van den Brand M, Hebeda KM, Groenen PJ, van Krieken JH, Scheijen B (December 2017). "Novel developments in the pathogenesis and diagnosis of extranodal marginal zone lymphoma". Journal of Hematopathology. 10 (3–4): 91–107. doi:10.1007/s12308-017-0302-2. PMC 5712330. PMID 29225710.
- Violeta Filip P, Cuciureanu D, Sorina Diaconu L, Maria Vladareanu A, Silvia Pop C (2018). "MALT lymphoma: epidemiology, clinical diagnosis and treatment". Journal of Medicine and Life. 11 (3): 187–193. doi:10.25122/jml-2018-0035. PMC 6197515. PMID 30364585.
- Bento-Miranda M, Figueiredo C (December 2014). "Helicobacter heilmannii sensu lato: an overview of the infection in humans". World Journal of Gastroenterology. 20 (47): 17779–87. doi:10.3748/wjg.v20.i47.17779. PMC 4273128. PMID 25548476.
- Teixeira Mendes LS, Wotherspoon A (2017). "Marginal zone lymphoma: Associated autoimmunity and auto-immune disorders". Best Practice & Research. Clinical Haematology. 30 (1–2): 65–76. doi:10.1016/j.beha.2016.07.006. PMID 28288719.
- Ponzoni M, Ferreri AJ (2017). "Bacteria associated with marginal zone lymphomas". Best Practice & Research. Clinical Haematology. 30 (1–2): 32–40. doi:10.1016/j.beha.2017.01.001. PMID 28288714.
- Matutes E, Montalban C (2017). "Clinical features and management of non-gastrointestinal non-ocular extranodal mucosa associated lymphoid tissue (ENMALT) marginal zone lymphomas". Best Practice & Research. Clinical Haematology. 30 (1–2): 99–108. doi:10.1016/j.beha.2016.07.005. PMID 28288723.
- Sammassimo S, Pruneri G, Andreola G, Montoro J, Steffanoni S, Nowakowski GS, Gandini S, Negri M, Habermann TM, Raderer M, Li ZM, Zinzani PL, Adam P, Zucca E, Martinelli G (December 2016). "A retrospective international study on primary extranodal marginal zone lymphoma of the lung (BALT lymphoma) on behalf of International Extranodal Lymphoma Study Group (IELSG)". Hematological Oncology. 34 (4): 177–183. doi:10.1002/hon.2243. PMID 26152851.
- Pileri S, Ponzoni M (2017). "Pathology of nodal marginal zone lymphomas". Best Practice & Research. Clinical Haematology. 30 (1–2): 50–55. doi:10.1016/j.beha.2016.11.001. PMID 28288717.
- Olszewska-Szopa M, Wróbel T (August 2019). "Gastrointestinal non-Hodgkin lymphomas". Advances in Clinical and Experimental Medicine. 28 (8): 1119–1124. doi:10.17219/acem/94068. PMID 31414733.
- Dou W, Li J, Xu L, Zhu J, Hu K, Sui Z, Wang J, Xu L, Wang S, Yin G (September 2016). "Halitosis and helicobacter pylori infection: A meta-analysis". Medicine. 95 (39): e4223. doi:10.1097/MD.0000000000004223. PMC 5265885. PMID 27684792.
- Diaconu S, Predescu A, Moldoveanu A, Pop CS, Fierbințeanu-Braticevici C (2017). "Helicobacter pylori infection: old and new". Journal of Medicine and Life. 10 (2): 112–117. PMC 5467250. PMID 28616085.
- Strand DS, Kim D, Peura DA (January 2017). "25 Years of Proton Pump Inhibitors: A Comprehensive Review". Gut and Liver. 11 (1): 27–37. doi:10.5009/gnl15502. PMC 5221858. PMID 27840364.
- Bianchi G, Sohani AR (January 2018). "Heavy Chain Disease of the Small Bowel". Current Gastroenterology Reports. 20 (1): 3. doi:10.1007/s11894-018-0608-y. PMID 29372346.
- Zucca E, Bertoni F (April 2016). "The spectrum of MALT lymphoma at different sites: biological and therapeutic relevance". Blood. 127 (17): 2082–92. doi:10.1182/blood-2015-12-624304. PMID 26989205.
- Won JH, Kim SM, Kim JW, Park JH, Kim JY (2019). "Clinical features, treatment and outcomes of colorectal mucosa-associated lymphoid tissue (MALT) lymphoma: literature reviews published in English between 1993 and 2017". Cancer Management and Research. 11: 8577–8587. doi:10.2147/CMAR.S214197. PMC 6759223. PMID 31572011.
- Takenaka R, Tomoda J, Sakata T, Ichiba T, Motoi M, Mizuno M, Tsuji T (March 2000). "Mucosa-associated lymphoid tissue lymphoma of the rectum that regressed spontaneously". Journal of Gastroenterology and Hepatology. 15 (3): 331–5. doi:10.1046/j.1440-1746.2000.02086.x. PMID 10764039.
- Kobayashi T, Takahashi N, Hagiwara Y, Tamaru J, Kayano H, Jin-nai I, Bessho M, Niitsu N (January 2008). "Successful radiotherapy in a patient with primary rectal mucosa-associated lymphoid tissue lymphoma without the API2-MALT1 fusion gene: a case report and review of the literature". Leukemia Research. 32 (1): 173–5. doi:10.1016/j.leukres.2007.04.017. PMID 17570523.
- Ma Q, Zhang C, Fang S, Zhong P, Zhu X, Lin L, Xiao H (March 2017). "Primary esophageal mucosa-associated lymphoid tissue lymphoma: A case report and review of literature". Medicine. 96 (13): e6478. doi:10.1097/MD.0000000000006478. PMC 5380272. PMID 28353588.
- Tabibian JH, Kalani A, Moran AM, Panganamamula K (January 2019). "Extranodal Marginal Zone B Cell (MALT) Lymphoma of the Esophagus". Journal of Gastrointestinal Cancer. doi:10.1007/s12029-018-00187-5. PMID 30618000.
- Takeuchi Y, Miyahara K, Morito T, Okikawa Y, Kinugasa H, Moritou Y, Higashi R, Kunihiro M, Nakagawa M (January 2019). "The Progression of Esophageal Mucosa-associated Lymphoid Tissue Lymphoma after Helicobacter pylori Eradication Therapy: A Case Report and Discussion of Therapeutic Options". Internal Medicine (Tokyo, Japan). 58 (2): 207–212. doi:10.2169/internalmedicine.1112-18. PMC 6378146. PMID 30146577.
- Sassone M, Ponzoni M, Ferreri AJ (2017). "Ocular adnexal marginal zone lymphoma: Clinical presentation, pathogenesis, diagnosis, prognosis, and treatment". Best Practice & Research. Clinical Haematology. 30 (1–2): 118–130. doi:10.1016/j.beha.2016.11.002. PMID 28288706.
- Tanenbaum RE, Galor A, Dubovy SR, Karp CL (2019). "Classification, diagnosis, and management of conjunctival lymphoma". Eye and Vision (London, England). 6: 22. doi:10.1186/s40662-019-0146-1. PMC 6660942. PMID 31372366.
- Guffey Johnson J, Terpak LA, Margo CE, Setoodeh R (April 2016). "Extranodal Marginal Zone B-cell Lymphoma of the Ocular Adnexa". Cancer Control : Journal of the Moffitt Cancer Center. 23 (2): 140–9. doi:10.1177/107327481602300208. PMID 27218791.
- Kempf W, Zimmermann AK, Mitteldorf C (June 2019). "Cutaneous lymphomas-An update 2019". Hematological Oncology. 37 Suppl 1: 43–47. doi:10.1002/hon.2584. PMID 31187534.
- Wilcox RA (November 2018). "Cutaneous B-cell lymphomas: 2019 update on diagnosis, risk stratification, and management". American Journal of Hematology. 93 (11): 1427–1430. doi:10.1002/ajh.25224. hdl:2027.42/109952. PMID 30039522.
- Ferreri AJ, Govi S, Ponzoni M (December 2013). "Marginal zone lymphomas and infectious agents". Seminars in Cancer Biology. 23 (6): 431–40. doi:10.1016/j.semcancer.2013.09.004. PMID 24090976.
- Ponzoni M, Ferreri AJ, Mappa S, Pasini E, Govi S, Facchetti F, Fanoni D, Tucci A, Vino A, Doglioni C, Berti E, Dolcetti R (2011). "Prevalence of Borrelia burgdorferi infection in a series of 98 primary cutaneous lymphomas". The Oncologist. 16 (11): 1582–8. doi:10.1634/theoncologist.2011-0108. PMC 3233293. PMID 22071292.
- Borie R, Wislez M, Antoine M, Cadranel J (2017). "Lymphoproliferative Disorders of the Lung". Respiration; International Review of Thoracic Diseases. 94 (2): 157–175. doi:10.1159/000477740. PMID 28609772.
- Defrancesco I, Arcaini L (March 2018). "Overview on the management of non-gastric MALT lymphomas". Best Practice & Research. Clinical Haematology. 31 (1): 57–64. doi:10.1016/j.beha.2017.11.001. PMID 29452667.
- Ahmed R, Al-Shaikh S, Akhtar M (May 2012). "Hashimoto thyroiditis: a century later". Advances in Anatomic Pathology. 19 (3): 181–6. doi:10.1097/PAP.0b013e3182534868. PMID 22498583.
- Stein SA, Wartofsky L (August 2013). "Primary thyroid lymphoma: a clinical review". The Journal of Clinical Endocrinology and Metabolism. 98 (8): 3131–8. doi:10.1210/jc.2013-1428. PMID 23714679.
- Watanabe N, Narimatsu H, Noh JY, Iwaku K, Kunii Y, Suzuki N, Ohye H, Suzuki M, Matsumoto M, Yoshihara A, Kameyama K, Kobayashi K, Kami M, Sugino K, Ito K (February 2018). "Long-Term Outcomes of 107 Cases of Primary Thyroid Mucosa-Associated Lymphoid Tissue Lymphoma at a Single Medical Institution in Japan". The Journal of Clinical Endocrinology and Metabolism. 103 (2): 732–739. doi:10.1210/jc.2017-01478. PMID 29165612.
- Peixoto R, Correia Pinto J, Soares V, Koch P, Taveira Gomes A (January 2017). "Primary thyroid lymphoma: A case report and review of the literature". Annals of Medicine and Surgery (2012). 13: 29–33. doi:10.1016/j.amsu.2016.12.023. PMC 5199157. PMID 28053701.
- Kleinstern G, Averbuch M, Abu Seir R, Perlman R, Ben Yehuda D, Paltiel O (April 2018). "Presence of autoimmune disease affects not only risk but also survival in patients with B-cell non-Hodgkin lymphoma". Hematological Oncology. 36 (2): 457–462. doi:10.1002/hon.2498. PMID 29469175.
- Dasanu CA, Bockorny B, Grabska J, Codreanu I (April 2015). "Prevalence and Pattern of Autoimmune Conditions in Patients with Marginal Zone Lymphoma: A Single Institution Experience". Connecticut Medicine. 79 (4): 197–200. PMID 26259295.
- Ayanambakkam A, Ibrahimi S, Bilal K, Cherry MA (January 2018). "Extranodal Marginal Zone Lymphoma of the Central Nervous System". Clinical Lymphoma, Myeloma & Leukemia. 18 (1): 34–37.e8. doi:10.1016/j.clml.2017.09.012. PMID 29103980.
- Razaq W, Goel A, Amin A, Grossbard ML (June 2009). "Primary central nervous system mucosa-associated lymphoid tissue lymphoma: case report and literature review". Clinical Lymphoma & Myeloma. 9 (3): E5–9. doi:10.3816/CLM.2009.n.052. PMID 19525185.
- Hissourou Iii M, Zia SY, Alqatari M, Strauchen J, Bakst RL (2016). "Primary MALT Lymphoma of the Breast Treated with Definitive Radiation". Case Reports in Hematology. 2016: 1831792. doi:10.1155/2016/1831792. PMC 4877461. PMID 27247809.
- Liguori G, Cantile M, Cerrone M, La Mantia E, Di Bonito M, Zanconati F, Curcio MP, Aquino G, La Mura A, Giannatiempo R, De Chiara A, Lombardi A, Botti G, D'Antonio A, Caraglia M, Franco R (October 2012). "Breast MALT lymphomas: a clinicopathological and cytogenetic study of 9 cases". Oncology Reports. 28 (4): 1211–6. doi:10.3892/or.2012.1942. PMID 22842723.
- Vempati P, Knoll MA, Alqatari M, Strauchen J, Malone AK, Bakst RL (2015). "MALT Lymphoma of the Bladder: A Case Report and Review of the Literature". Case Reports in Hematology. 2015: 934374. doi:10.1155/2015/934374. PMC 4568333. PMID 26417464.
- Venyo AK (2014). "Lymphoma of the urinary bladder". Advances in Urology. 2014: 327917. doi:10.1155/2014/327917. PMC 3912819. PMID 24511310.
- Garcia M, Konoplev S, Morosan C, Abruzzo LV, Bueso-Ramos CE, Medeiros LJ (September 2007). "MALT lymphoma involving the kidney: a report of 10 cases and review of the literature". American Journal of Clinical Pathology. 128 (3): 464–73. doi:10.1309/0T2UKUKV91W3QR6W. PMID 17709321.
- Makino T, Miwa S, Koshida K, Kawashima A (April 2016). "Mucosa-associated lymphoid tissue lymphoma involving the kidney: a case report and review of the literature". International Cancer Conference Journal. 5 (2): 82–89. doi:10.1007/s13691-015-0234-6. PMC 6498318. PMID 31149432.
- Mani H, Climent F, Colomo L, Pittaluga S, Raffeld M, Jaffe ES (September 2010). "Gall bladder and extrahepatic bile duct lymphomas: clinicopathological observations and biological implications". The American Journal of Surgical Pathology. 34 (9): 1277–86. doi:10.1097/PAS.0b013e3181e9bb8b. PMC 2929270. PMID 20679881.
- Honda M, Furuta Y, Naoe H, Sasaki Y (May 2017). "Primary mucosa-associated lymphoid tissue (MALT) lymphoma of the gallbladder and review of the literature". BMJ Case Reports. 2017. doi:10.1136/bcr-2017-220161. PMC 5747624. PMID 28551602.
- Bohlok A, De Grez T, Bouazza F, De Wind R, El-Khoury M, Repullo D, Donckier V (2018). "Primary Hepatic Lymphoma Mimicking a Hepatocellular Carcinoma in a Cirrhotic Patient: Case Report and Systematic Review of the Literature". Case Reports in Surgery. 2018: 9183717. doi:10.1155/2018/9183717. PMC 5914115. PMID 29850362.
- Xie H, Lv J, Ji Y, Du X, Yang X (March 2019). "Primary hepatic mucosa-associated lymphoid tissue lymphoma: A case report and literature review". Medicine. 98 (13): e15034. doi:10.1097/MD.0000000000015034. PMC 6456129. PMID 30921228.
- Armand M, Besson C, Hermine O, Davi F (2017). "Hepatitis C virus - Associated marginal zone lymphoma". Best Practice & Research. Clinical Haematology. 30 (1–2): 41–49. doi:10.1016/j.beha.2017.02.001. PMID 28288715.
- Mihăilă RG (July 2016). "Hepatitis C virus - associated B cell non-Hodgkin's lymphoma". World Journal of Gastroenterology. 22 (27): 6214–23. doi:10.3748/wjg.v22.i27.6214. PMC 4945980. PMID 27468211.
- Jaramillo Oquendo C, Parker H, Oscier D, Ennis S, Gibson J, Strefford JC (July 2019). "Systematic Review of Somatic Mutations in Splenic Marginal Zone Lymphoma". Scientific Reports. 9 (1): 10444. Bibcode:2019NatSR...910444J. doi:10.1038/s41598-019-46906-1. PMC 6639539. PMID 31320741.
- Arcaini L, Rossi D, Paulli M (April 2016). "Splenic marginal zone lymphoma: from genetics to management". Blood. 127 (17): 2072–81. doi:10.1182/blood-2015-11-624312. PMID 26989207.
- Piris MA, Onaindía A, Mollejo M (2017). "Splenic marginal zone lymphoma". Best Practice & Research. Clinical Haematology. 30 (1–2): 56–64. doi:10.1016/j.beha.2016.09.005. PMID 28288718.
- Kalpadakis C, Pangalis GA, Angelopoulou MK, Vassilakopoulos TP (2017). "Treatment of splenic marginal zone lymphoma". Best Practice & Research. Clinical Haematology. 30 (1–2): 139–148. doi:10.1016/j.beha.2016.07.004. PMID 28288709.
- Spina V, Rossi D (2017). "Molecular pathogenesis of splenic and nodal marginal zone lymphoma". Best Practice & Research. Clinical Haematology. 30 (1–2): 5–12. doi:10.1016/j.beha.2016.09.004. PMID 28288716.
- Jha P, Das H (November 2017). "KLF2 in Regulation of NF-κB-Mediated Immune Cell Function and Inflammation". International Journal of Molecular Sciences. 18 (11): 2383. doi:10.3390/ijms18112383. PMC 5713352. PMID 29125549.
- Sakata-Yanagimoto M, Chiba S (2012). "Notch2 and immune function". In Radtke F (ed.). Notch Regulation of the Immune System. Current Topics in Microbiology and Immunology. 360. pp. 151–61. doi:10.1007/82_2012_235. ISBN 978-3-642-24293-9. PMID 22695918.
- Iannitto E, Bellei M, Amorim S, Ferreri AJ, Marcheselli L, Cesaretti M, Haioun C, Mancuso S, Bouabdallah K, Gressin R, Tripodo C, Traverse-Glehen A, Baseggio L, Zupo S, Stelitano C, Castagnari B, Patti C, Alvarez I, Liberati AM, Merli M, Gini G, Cabras MG, Dupuis J, Tessoulin B, Perrot A, Re F, Palombi F, Gulino A, Zucca E, Federico M, Thieblemont C (December 2018). "Efficacy of bendamustine and rituximab in splenic marginal zone lymphoma: results from the phase II BRISMA/IELSG36 study". British Journal of Haematology. 183 (5): 755–765. doi:10.1111/bjh.15641. PMID 30407629.
- Thieblemont C, Molina T, Davi F (April 2016). "Optimizing therapy for nodal marginal zone lymphoma". Blood. 127 (17): 2064–71. doi:10.1182/blood-2015-12-624296. PMID 26989202.
- Qu Q, Xuan W, Fan GH (January 2015). "Roles of resolvins in the resolution of acute inflammation". Cell Biology International. 39 (1): 3–22. doi:10.1002/cbin.10345. PMID 25052386.
- Tadmor T, Polliack A (2017). "Nodal marginal zone lymphoma: Clinical features, diagnosis, management and treatment". Best Practice & Research. Clinical Haematology. 30 (1–2): 92–98. doi:10.1016/j.beha.2016.08.026. PMID 28288722.
- van den Brand M, van Krieken JH (July 2013). "Recognizing nodal marginal zone lymphoma: recent advances and pitfalls. A systematic review". Haematologica. 98 (7): 1003–13. doi:10.3324/haematol.2012.083386. PMC 3696602. PMID 23813646.
- Rudolf R, Busch R, Patra AK, Muhammad K, Avots A, Andrau JC, Klein-Hessling S, Serfling E (2014). "Architecture and expression of the nfatc1 gene in lymphocytes". Frontiers in Immunology. 5: 21. doi:10.3389/fimmu.2014.00021. PMC 3909943. PMID 24550910.
- Brown A, Sciascia-Visani I, Farrell D, Smith M, Felix C, Mutharajah V, Ruell J, Taylor G (2019). "A patient with a diagnosis of nodal marginal zone B-cell lymphoma and a t(2;14)(p24;q32) involving MYCN and IGH". Molecular Cytogenetics. 12: 3. doi:10.1186/s13039-019-0419-3. PMC 6359751. PMID 30733831.
- Woessmann W, Quintanilla-Martinez L (June 2019). "Rare mature B-cell lymphomas in children and adolescents". Hematological Oncology. 37 Suppl 1: 53–61. doi:10.1002/hon.2585. PMID 31187530.
- "Marginal Zone Lymphoma - Lymphoma Research Foundation". www.lymphoma.org. Retrieved 2017-12-10.
- "FDA Approves First-Ever Targeted Marginal Zone Lymphoma Treatment". New Developments in Lymphoma. 2017-01-19. Retrieved 2017-12-10.
- Blosse A, Lehours P, Wilson KT, Gobert AP (September 2018). "Helicobacter: Inflammation, immunology, and vaccines". Helicobacter. 23 Suppl 1: e12517. doi:10.1111/hel.12517. PMC 6310010. PMID 30277626.
External links
| Classification |
|---|