Glutathione S-transferase
Glutathione S-transferases (GSTs), previously known as ligandins, comprise a family of eukaryotic and prokaryotic phase II metabolic isozymes best known for their ability to catalyze the conjugation of the reduced form of glutathione (GSH) to xenobiotic substrates for the purpose of detoxification. The GST family consists of three superfamilies: the cytosolic, mitochondrial, and microsomal—also known as MAPEG—proteins.[1][2][3] Members of the GST superfamily are extremely diverse in amino acid sequence, and a large fraction of the sequences deposited in public databases are of unknown function.[4] The Enzyme Function Initiative (EFI) is using GSTs as a model superfamily to identify new GST functions.
| Glutathione S-transferase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
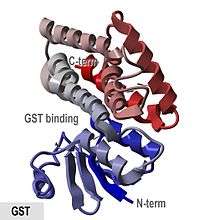 Crystallographic structure of glutathione S-transferase from Anopheles cracens.[1] | |||||||||
| Identifiers | |||||||||
| EC number | 2.5.1.18 | ||||||||
| CAS number | 50812-37-8 | ||||||||
| Databases | |||||||||
| IntEnz | IntEnz view | ||||||||
| BRENDA | BRENDA entry | ||||||||
| ExPASy | NiceZyme view | ||||||||
| KEGG | KEGG entry | ||||||||
| MetaCyc | metabolic pathway | ||||||||
| PRIAM | profile | ||||||||
| PDB structures | RCSB PDB PDBe PDBsum | ||||||||
| Gene Ontology | AmiGO / QuickGO | ||||||||
| |||||||||
GSTs can constitute up to 10% of cytosolic protein in some mammalian organs.[5][6] GSTs catalyse the conjugation of GSH—via a sulfhydryl group—to electrophilic centers on a wide variety of substrates in order to make the compounds more water-soluble.[7][8] This activity detoxifies endogenous compounds such as peroxidised lipids and enables the breakdown of xenobiotics. GSTs may also bind toxins and function as transport proteins, which gave rise to the early term for GSTs, ligandin.[9][10]
Classification
| Glutathione S-transferase, C-terminal domain | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 Structure of the xenobiotic substrate binding site of rat glutathione S-transferase mu 1 bound to the GSH adduct of phenanthrene-9,10-oxide.[11] | |||||||||
| Identifiers | |||||||||
| Symbol | GST_C | ||||||||
| Pfam | PF00043 | ||||||||
| InterPro | IPR004046 | ||||||||
| SCOPe | 2gst / SUPFAM | ||||||||
| OPM superfamily | 178 | ||||||||
| OPM protein | 5i9k | ||||||||
| CDD | cd00299 | ||||||||
| |||||||||
Protein sequence and structure are important additional classification criteria for the three superfamilies (cytosolic, mitochondrial, and MAPEG) of GSTs: while classes from the cytosolic superfamily of GSTs possess more than 40% sequence homology, those from other classes may have less than 25%. Cytosolic GSTs are divided into 13 classes based upon their structure: alpha, beta, delta, epsilon, zeta, theta, mu, nu, pi, sigma, tau, phi, and omega. Mitochondrial GSTs are in class kappa. The MAPEG superfamily of microsomal GSTs consists of subgroups designated I-IV, between which amino acid sequences share less than 20% identity. Human cytosolic GSTs belong to the alpha, zeta, theta, mu, pi, sigma, and omega classes, while six isozymes belonging to classes I, II, and IV of the MAPEG superfamily are known to exist.[8][12][13]
Nomenclature
Standardized GST nomenclature first proposed in 1992 identifies the species to which the isozyme of interest belongs with a lower-case initial (e.g., "h" for human), which precedes the abbreviation GST. The isozyme class is subsequently identified with an upper-case letter (e.g., "A" for alpha), followed by an Arabic numeral representing the class subfamily (or subunit). Because both mitochondrial and cytosolic GSTs exist as dimers, and only heterodimers form between members of the same class, the second subfamily component of the enzyme dimer is denoted with a hyphen, followed by an additional Arabic numeral.[12][13] Therefore, if a human glutathione S-transferase is a homodimer in the pi-class subfamily 1, its name will be written as "hGSTP1-1."
The early nomenclature for GSTs referred to them as “Y” proteins, referring to their separation in the “Y” fraction (as opposed to the “X and Z” fractions) using Sephadex G75 chromatography.[14] As GST sub-units were identified they were referred to as Ya, Yp, etc. with if necessary, a number identifying the monomer isoform (e.g. Yb1). Litwack et al proposed the term “Ligandin” to cover the proteins previously known as “Y” proteins.[10]
In clinical chemistry and toxicology, the terms alpha GST, mu GST, and pi GST are most commonly used.
Structure
| Identifiers | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Symbol | GST_N | ||||||||
| Pfam | PF02798 | ||||||||
| Pfam clan | CL0172 | ||||||||
| InterPro | IPR004045 | ||||||||
| PROSITE | PS50404 | ||||||||
| SCOPe | 1g7o / SUPFAM | ||||||||
| |||||||||
The glutathione binding site, or "G-site", is located in the thioredoxin-like domain of both cytosolic and mitochondrial GSTs. The region containing the greatest amount of variability between the assorted classes is that of helix α2, where one of three different amino acid residues interacts with the glycine residue of glutathione. Two subgroups of cytosolic GSTs have been characterized based upon their interaction with glutathione: the Y-GST group, which uses a tyrosine residue to activate glutathione, and the S/C-GST, which instead uses serine or cysteine residues.[8][15]
- "GST proteins are globular proteins with an N-terminal mixed helical and beta-strand domain and an all-helical C-terminal domain."
The porcine pi-class enzyme pGTSP1-1 was the first GST to have its structure determined, and it is representative of other members of the cytosolic GST superfamily, which contain a thioredoxin-like N-terminal domain as well as a C-terminal domain consisting of alpha helices.[8][16]
Mammalian cytosolic GSTs are dimeric, with both subunits being from the same class of GSTs, although not necessarily identical. The monomers are approximately 25 kDa in size.[12][17] They are active over a wide variety of substrates with considerable overlap.[18] The following table lists all GST enzymes of each class known to exist in Homo sapiens, as found in the UniProtKB/Swiss-Prot database.
| GST Class | Homo sapiens GST Class Members (22) |
|---|---|
| Alpha | GSTA1, GSTA2, GSTA3, GSTA4, GSTA5 |
| Delta | |
| Kappa | GSTK1 |
| Mu | GSTM1, GSTM1L (RNAi), GSTM2, GSTM3, GSTM4, GSTM5 |
| Omega | GSTO1, GSTO2 |
| Pi | GSTP1 |
| Theta | GSTT1, GSTT2, GSTT4 |
| Zeta | GSTZ1 (aka MAAI-Maleylacetoacetate isomerase) |
| Microsomal | MGST1, MGST2, MGST3 |
Function
The activity of GSTs is dependent upon a steady supply of GSH from the synthetic enzymes gamma-glutamylcysteine synthetase and glutathione synthetase, as well as the action of specific transporters to remove conjugates of GSH from the cell. The primary role of GSTs is to detoxify xenobiotics by catalyzing the nucleophilic attack by GSH on electrophilic carbon, sulfur, or nitrogen atoms of said nonpolar xenobiotic substrates, thereby preventing their interaction with crucial cellular proteins and nucleic acids.[13][19] Specifically, the function of GSTs in this role is twofold: to bind both the substrate at the enzyme's hydrophobic H-site and GSH at the adjacent, hydrophilic G-site, which together form the active site of the enzyme; and subsequently to activate the thiol group of GSH, enabling the nucleophilic attack upon the substrate.[12] The glutathione molecule binds in a cleft between N- and C-terminal domains - the catalytically important residues are proposed to reside in the N-terminal domain.[20] Both subunits of the GST dimer, whether hetero- or homodimeric in nature, contain a single nonsubstrate binding site, as well as a GSH-binding site. In heterodimeric GST complexes such as those formed by the cytosolic mu and alpha classes, however, the cleft between the two subunits is home to an additional high-affinity nonsubstrate xenobiotic binding site, which may account for the enzymes' ability to form heterodimers.[19][21]
The compounds targeted in this manner by GSTs encompass a diverse range of environmental or otherwise exogenous toxins, including chemotherapeutic agents and other drugs, pesticides, herbicides, carcinogens, and variably-derived epoxides; indeed, GSTs are responsible for the conjugation of β1-8,9-epoxide, a reactive intermediate formed from aflatoxin B1, which is a crucial means of protection against the toxin in rodents. The detoxification reactions comprise the first four steps of mercapturic acid synthesis,[19] with the conjugation to GSH serving to make the substrates more soluble and allowing them to be removed from the cell by transporters such as multidrug resistance-associated protein 1 (MRP1).[8] After export, the conjugation products are converted into mercapturic acids and excreted via the urine or bile.[13]
Most mammalian isoenzymes have affinity for the substrate 1-chloro-2,4-dinitrobenzene, and spectrophotometric assays utilising this substrate are commonly used to report GST activity.[22] However, some endogenous compounds, e.g., bilirubin, can inhibit the activity of GSTs. In mammals, GST isoforms have cell specific distributions (for example, α-GST in hepatocytes and π-GST in the biliary tract of the human liver).[23]
Role in cell signaling
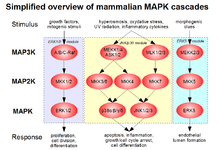
Although best known for their ability to conjugate xenobiotics to GSH and thereby detoxify cellular environments, GSTs are also capable of binding nonsubstrate ligands, with important cell signaling implications. Several GST isozymes from various classes have been shown to inhibit the function of a kinase involved in the MAPK pathway that regulates cell proliferation and death, preventing the kinase from carrying out its role in facilitating the signaling cascade.[24]
Cytosolic GSTP1-1, a well-characterized isozyme of the mammalian GST family, is expressed primarily in heart, lung, and brain tissues; in fact, it is the most common GST expressed outside the liver.[24][25] Based on its overexpression in a majority of human tumor cell lines and prevalence in chemotherapeutic-resistant tumors, GSTP1-1 is thought to play a role in the development of cancer and its potential resistance to drug treatment. Further evidence for this comes from the knowledge that GSTP can selectively inhibit C-Jun phosphorylation by JNK, preventing apoptosis.[24] During times of low cellular stress, a complex forms through direct protein–protein interactions between GSTP and the C-terminus of JNK, effectively preventing the action of JNK and thus its induction of the JNK pathway. Cellular oxidative stress causes the dissociation of the complex, oligomerization of GSTP, and induction of the JNK pathway, resulting in apoptosis.[26] The connection between GSTP inhibition of the pro-apoptotic JNK pathway and the isozyme's overexpression in drug-resistant tumor cells may itself account for the tumor cells' ability to escape apoptosis mediated by drugs that are not substrates of GSTP.[24]
Like GSTP, GSTM1 is involved in regulating apoptotic pathways through direct protein–protein interactions, although it acts on ASK1, which is upstream of JNK. The mechanism and result are similar to that of GSTP and JNK, in that GSTM1 sequesters ASK1 through complex formation and prevents its induction of the pro-apoptotic p38 and JNK portions of the MAPK signaling cascade. Like GSTP, GSTM1 interacts with its partner in the absence of oxidative stress, although ASK1 is also involved in heat shock response, which is likewise prevented during ASK1 sequestration. The fact that high levels of GST are associated with resistance to apoptosis induced by a range of substances, including chemotherapeutic agents, supports its putative role in MAPK signaling prevention.[26]
Implications in cancer development
There is a growing body of evidence supporting the role of GST, particularly GSTP, in cancer development and chemotherapeutic resistance. The link between GSTP and cancer is most obvious in the overexpression of GSTP in many cancers, but it is also supported by the fact that the transformed phenotype of tumor cells is associated with aberrantly regulated kinase signaling pathways and cellular addiction to overexpressed proteins. That most anti-cancer drugs are poor substrates for GSTP indicates that the role of elevated GSTP in many tumor cell lines is not to detoxify the compounds, but must have another purpose; this hypothesis is also given credence by the common finding of GSTP overexpression in tumor cell lines that are not drug resistant.[27]
Clinical significance
In addition to their roles in cancer development and chemotherapeutic drug resistance, GSTs are implicated in a variety of diseases by virtue of their involvement with GSH. Although the evidence is minimal for the influence of GST polymorphisms of the alpha, mu, pi, and theta classes on susceptibility to various types of cancer, numerous studies have implicated such genotypic variations in asthma, atherosclerosis, allergies, and other inflammatory diseases.[19]
Because diabetes is a disease that involves oxidative damage, and GSH metabolism is dysfunctional in diabetic patients, GSTs may represent a potential target for diabetic drug treatment. In addition, insulin administration is known to result in increased GST gene expression through the PI3K/AKT/mTOR pathway and reduced intracellular oxidative stress, while glucagon decreases such gene expression.[28]
Omega-class GST (GSTO) genes, in particular, are associated with neurological diseases such as Alzheimer's, Parkinson's, and amyotrophic lateral sclerosis; again, oxidative stress is believed to be the culprit, with decreased GSTO gene expression resulting in a lowered age of onset for the diseases.[29]
Release of GSTs as an indication of organ damage
The high intracellular concentrations of GSTs coupled with their cell-specific cellular distribution allows them to function as biomarkers for localising and monitoring injury to defined cell types. For example, hepatocytes contain high levels of alpha GST and serum alpha GST has been found to be an indicator of hepatocyte injury in transplantation, toxicity and viral infections.[30][31][32]
Similarly, in humans, renal proximal tubular cells contain high concentrations of alpha GST, while distal tubular cells contain pi GST.[33] This specific distribution enables the measurement of urinary GSTs to be used to quantify and localise renal tubular injury in transplantation, nephrotoxicity and ischaemic injury.[34]
In rodent pre-clinical studies, urinary and serum alpha GST have been shown to be sensitive and specific indicators of renal proximal tubular and hepatocyte necrosis respectively.[35][36]
GST-tags and the GST pull-down assay
GST can be added to a protein of interest to purify it from solution in a process known as a pull-down assay. This is accomplished by inserting the GST DNA coding sequence next to that which codes for the protein of interest. Thus, after transcription and translation, the GST protein and the protein of interest will be expressed together as a fusion protein. Because the GST protein has a strong binding affinity for GSH, beads coated with the compound can be added to the protein mixture; as a result, the protein of interest attached to the GST will stick to the beads, isolating the protein from the rest of those in solution. The beads are recovered and washed with free GSH to detach the protein of interest from the beads, resulting in a purified protein. This technique can be used to elucidate direct protein–protein interactions. A drawback of this assay is that the protein of interest is attached to GST, altering its native state.[37][38]
A GST-tag is often used to separate and purify proteins that contain the GST-fusion protein. The tag is 220 amino acids (roughly 26 kDa) in size,[39] which, compared to tags such as the Myc-tag or the FLAG-tag, is quite large. It can be fused to either the N-terminus or C-terminus of a protein. However, many commercially available sources of GST-tagged plasmids include a thrombin domain for cleavage of the GST tag during protein purification.[37][40]
See also
References
- PDB: 1R5A; Udomsinprasert R, Pongjaroenkit S, Wongsantichon J, Oakley AJ, Prapanthadara LA, Wilce MC, Ketterman AJ (June 2005). "Identification, characterization and structure of a new Delta class glutathione transferase isoenzyme". The Biochemical Journal. 388 (Pt 3): 763–71. doi:10.1042/BJ20042015. PMC 1183455. PMID 15717864.
- Sheehan D, Meade G, Foley VM, Dowd CA (November 2001). "Structure, function and evolution of glutathione transferases: implications for classification of non-mammalian members of an ancient enzyme superfamily". The Biochemical Journal. 360 (Pt 1): 1–16. doi:10.1042/0264-6021:3600001. PMC 1222196. PMID 11695986.
- Allocati N, Federici L, Masulli M, Di Ilio C (January 2009). "Glutathione transferases in bacteria". The FEBS Journal. 276 (1): 58–75. doi:10.1111/j.1742-4658.2008.06743.x. PMID 19016852.
- Atkinson HJ, Babbitt PC (November 2009). "Glutathione transferases are structural and functional outliers in the thioredoxin fold". Biochemistry. 48 (46): 11108–16. doi:10.1021/bi901180v. PMC 2778357. PMID 19842715.
- Boyer TD (March 1989). "The glutathione S-transferases: an update". Hepatology. 9 (3): 486–96. doi:10.1002/hep.1840090324. PMID 2646197.
- Mukanganyama S, Bezabih M, Robert M, Ngadjui BT, Kapche GF, Ngandeu F, Abegaz B (August 2011). "The evaluation of novel natural products as inhibitors of human glutathione transferase P1-1". Journal of Enzyme Inhibition and Medicinal Chemistry. 26 (4): 460–7. doi:10.3109/14756366.2010.526769. PMID 21028940.
- Douglas KT (1987). "Mechanism of action of glutathione-dependent enzymes". Advances in Enzymology and Related Areas of Molecular Biology. Advances in Enzymology and Related Areas of Molecular Biology. 59. pp. 103–67. doi:10.1002/9780470123058.ch3. ISBN 9780470123058. PMID 2880477.
- Oakley A (May 2011). "Glutathione transferases: a structural perspective". Drug Metabolism Reviews. 43 (2): 138–51. doi:10.3109/03602532.2011.558093. PMID 21428697.
- Leaver MJ, George SG (1998). "A piscine glutathione S-transferase which efficiently conjugates the end-products of lipid peroxidation". Marine Environmental Research. 46 (1–5): 71–74. doi:10.1016/S0141-1136(97)00071-8.
- Litwack G, Ketterer B, Arias IM (December 1971). "Ligandin: a hepatic protein which binds steroids, bilirubin, carcinogens and a number of exogenous organic anions". Nature. 234 (5330): 466–7. doi:10.1038/234466a0. PMID 4944188.
- PDB: 2GST; Ji X, Johnson WW, Sesay MA, Dickert L, Prasad SM, Ammon HL, Armstrong RN, Gilliland GL (February 1994). "Structure and function of the xenobiotic substrate binding site of a glutathione S-transferase as revealed by X-ray crystallographic analysis of product complexes with the diastereomers of 9-(S-glutathionyl)-10-hydroxy-9,10-dihydrophenanthrene". Biochemistry. 33 (5): 1043–52. doi:10.1021/bi00171a002. PMID 8110735.
- Eaton DL, Bammler TK (June 1999). "Concise review of the glutathione S-transferases and their significance to toxicology". Toxicological Sciences. 49 (2): 156–64. doi:10.1093/toxsci/49.2.156. PMID 10416260.
- Josephy PD (June 2010). "Genetic variations in human glutathione transferase enzymes: significance for pharmacology and toxicology". Human Genomics and Proteomics. 2010: 876940. doi:10.4061/2010/876940. PMC 2958679. PMID 20981235.
- Levi AJ, Gatmaitan Z, Arias IM (November 1969). "Two hepatic cytoplasmic protein fractions, Y and Z, and their possible role in the hepatic uptake of bilirubin, sulfobromophthalein, and other anions". The Journal of Clinical Investigation. 48 (11): 2156–67. doi:10.1172/JCI106182. PMC 297469. PMID 4980931.
- Atkinson HJ, Babbitt PC (November 2009). "Glutathione transferases are structural and functional outliers in the thioredoxin fold". Biochemistry. 48 (46): 11108–16. doi:10.1021/bi901180v. PMC 2778357. PMID 19842715.
- Park AK, Moon JH, Jang EH, Park H, Ahn IY, Lee KS, Chi YM (March 2013). "The structure of a shellfish specific GST class glutathione S-transferase from antarctic bivalve Laternula elliptica reveals novel active site architecture". Proteins. 81 (3): 531–7. doi:10.1002/prot.24208. PMID 23152139.
- Landi S (October 2000). "Mammalian class theta GST and differential susceptibility to carcinogens: a review". Mutation Research. 463 (3): 247–83. doi:10.1016/s1383-5742(00)00050-8. PMID 11018744.
- Raza H (November 2011). "Dual localization of glutathione S-transferase in the cytosol and mitochondria: implications in oxidative stress, toxicity and disease". The FEBS Journal. 278 (22): 4243–51. doi:10.1111/j.1742-4658.2011.08358.x. PMC 3204177. PMID 21929724.
- Hayes JD, Flanagan JU, Jowsey IR (2005). "Glutathione transferases". Annual Review of Pharmacology and Toxicology. 45: 51–88. doi:10.1146/annurev.pharmtox.45.120403.095857. PMID 15822171.
- Nishida M, Harada S, Noguchi S, Satow Y, Inoue H, Takahashi K (August 1998). "Three-dimensional structure of Escherichia coli glutathione S-transferase complexed with glutathione sulfonate: catalytic roles of Cys10 and His106". Journal of Molecular Biology. 281 (1): 135–47. doi:10.1006/jmbi.1998.1927. PMID 9680481.
- Vargo MA, Colman RF (January 2001). "Affinity labeling of rat glutathione S-transferase isozyme 1-1 by 17β-iodoacetoxy-estradiol-3-sulfate". The Journal of Biological Chemistry. 276 (3): 2031–6. doi:10.1074/jbc.M008212200. PMID 11031273.
- Habig WH, Pabst MJ, Fleischner G, Gatmaitan Z, Arias IM, Jakoby WB (October 1974). "The identity of glutathione S-transferase B with ligandin, a major binding protein of liver". Proceedings of the National Academy of Sciences of the United States of America. 71 (10): 3879–82. doi:10.1073/pnas.71.10.3879. PMC 434288. PMID 4139704.
- Beckett GJ, Hayes JD (1987). "Glutathione S-transferase measurements and liver disease in man". Journal of Clinical Biochemistry and Nutrition. 2: 1–24. doi:10.3164/jcbn.2.1.
- Laborde E (September 2010). "Glutathione transferases as mediators of signaling pathways involved in cell proliferation and cell death". Cell Death and Differentiation. 17 (9): 1373–80. doi:10.1038/cdd.2010.80. PMID 20596078.
- Adler V, Yin Z, Fuchs SY, Benezra M, Rosario L, Tew KD, Pincus MR, Sardana M, Henderson CJ, Wolf CR, Davis RJ, Ronai Z (March 1999). "Regulation of JNK signaling by GSTp". The EMBO Journal. 18 (5): 1321–34. doi:10.1093/emboj/18.5.1321. PMC 1171222. PMID 10064598.
- Townsend DM, Tew KD (October 2003). "The role of glutathione-S-transferase in anti-cancer drug resistance". Oncogene. 22 (47): 7369–75. doi:10.1038/sj.onc.1206940. PMC 6361125. PMID 14576844.
- Tew KD, Manevich Y, Grek C, Xiong Y, Uys J, Townsend DM (July 2011). "The role of glutathione S-transferase P in signaling pathways and S-glutathionylation in cancer". Free Radical Biology & Medicine. 51 (2): 299–313. doi:10.1016/j.freeradbiomed.2011.04.013. PMC 3125017. PMID 21558000.
- Franco R, Schoneveld OJ, Pappa A, Panayiotidis MI (2007). "The central role of glutathione in the pathophysiology of human diseases". Archives of Physiology and Biochemistry. 113 (4–5): 234–58. doi:10.1080/13813450701661198. PMID 18158646.
- Board PG (May 2011). "The omega-class glutathione transferases: structure, function, and genetics". Drug Metabolism Reviews. 43 (2): 226–35. doi:10.3109/03602532.2011.561353. PMID 21495794.
- Beckett GJ, Chapman BJ, Dyson EH, Hayes JD (January 1985). "Plasma glutathione S-transferase measurements after paracetamol overdose: evidence for early hepatocellular damage". Gut. 26 (1): 26–31. doi:10.1136/gut.26.1.26. PMC 1432412. PMID 3965363.
- Hughes VF, Trull AK, Gimson A, Friend PJ, Jamieson N, Duncan A, Wight DG, Prevost AT, Alexander GJ (November 1997). "Randomized trial to evaluate the clinical benefits of serum alpha-glutathione S-transferase concentration monitoring after liver transplantation". Transplantation. 64 (10): 1446–52. doi:10.1097/00007890-199711270-00013. PMID 9392310.
- Loguercio C, Caporaso N, Tuccillo C, Morisco F, Del Vecchio Blanco G, Del Vecchio Blanco C (March 1998). "Alpha-glutathione transferases in HCV-related chronic hepatitis: a new predictive index of response to interferon therapy?". Journal of Hepatology. 28 (3): 390–5. doi:10.1016/s0168-8278(98)80311-5. PMID 9551675.
- Harrison DJ, Kharbanda R, Cunningham DS, McLellan LI, Hayes JD (June 1989). "Distribution of glutathione S-transferase isoenzymes in human kidney: basis for possible markers of renal injury". Journal of Clinical Pathology. 42 (6): 624–8. doi:10.1136/jcp.42.6.624. PMC 1141991. PMID 2738168.
- Sundberg AG, Appelkvist EL, Bäckman L, Dallner G (1994). "Urinary pi-class glutathione transferase as an indicator of tubular damage in the human kidney". Nephron. 67 (3): 308–16. doi:10.1159/000187985. PMID 7936021.
- Harpur E, Ennulat D, Hoffman D, Betton G, Gautier JC, Riefke B, Bounous D, Schuster K, Beushausen S, Guffroy M, Shaw M, Lock E, Pettit S (August 2011). "Biological qualification of biomarkers of chemical-induced renal toxicity in two strains of male rat". Toxicological Sciences. 122 (2): 235–52. doi:10.1093/toxsci/kfr112. PMID 21593213.
- Bailey WJ, Holder D, Patel H, Devlin P, Gonzalez RJ, Hamilton V, Muniappa N, Hamlin DM, Thomas CE, Sistare FD, Glaab WE (December 2012). "A performance evaluation of three drug-induced liver injury biomarkers in the rat: alpha-glutathione S-transferase, arginase 1, and 4-hydroxyphenyl-pyruvate dioxygenase" (PDF). Toxicological Sciences. 130 (2): 229–44. doi:10.1093/toxsci/kfs243. PMID 22872058.
- Benard V, Bokoch GM (2002). Assay of Cdc42, Rac, and Rho GTPase activation by affinity methods. Methods in Enzymology. 345. pp. 349–59. doi:10.1016/s0076-6879(02)45028-8. ISBN 9780121822460. PMID 11665618.
- Ren L, Chang E, Makky K, Haas AL, Kaboord B, Walid Qoronfleh M (November 2003). "Glutathione S-transferase pull-down assays using dehydrated immobilized glutathione resin". Analytical Biochemistry. 322 (2): 164–9. doi:10.1016/j.ab.2003.07.023. PMID 14596823.
- Long F, Cho W, Ishii Y (September 2011). "Expression and purification of 15N- and 13C-isotope labeled 40-residue human Alzheimer's β-amyloid peptide for NMR-based structural analysis". Protein Expression and Purification. 79 (1): 16–24. doi:10.1016/j.pep.2011.05.012. PMC 3134129. PMID 21640828.
- Tinta T, Christiansen LS, Konrad A, Liberles DA, Turk V, Munch-Petersen B, Piškur J, Clausen AR (June 2012). "Deoxyribonucleoside kinases in two aquatic bacteria with high specificity for thymidine and deoxyadenosine". FEMS Microbiology Letters. 331 (2): 120–7. doi:10.1111/j.1574-6968.2012.02565.x. PMID 22462611.
External links
- Overview of Glutathione S-Transferases
- Glutathione+S-Transferase at the US National Library of Medicine Medical Subject Headings (MeSH)
- EC 2.5.1.18
- Preparation of GST Fusion Proteins
- GST Gene Fusion System Handbook