Glucose-6-phosphate dehydrogenase deficiency
Glucose-6-phosphate dehydrogenase deficiency (G6PDD) is an inborn error of metabolism that predisposes to red blood cell breakdown.[1] Most of the time, those who are affected have no symptoms.[3] Following a specific trigger, symptoms such as yellowish skin, dark urine, shortness of breath, and feeling tired may develop.[1][2] Complications can include anemia and newborn jaundice.[2] Some people never have symptoms.[3]
| Glucose-6-phosphate dehydrogenase deficiency | |
|---|---|
| Other names | Favism[1] |
 | |
| Glucose-6-phosphate dehydrogenase | |
| Specialty | Medical genetics |
| Symptoms | Yellowish skin, dark urine, shortness of breath[1] |
| Complications | Anemia, newborn jaundice[2][1] |
| Usual onset | Within a few days of a trigger[2] |
| Causes | Genetic (X-linked recessive)[1] |
| Risk factors | Triggered by infections, certain medication, stress, foods such as fava beans[1][3] |
| Diagnostic method | Based on symptoms, blood test, genetic testing[2] |
| Differential diagnosis | Pyruvate kinase deficiency, hereditary spherocytosis, sickle cell anemia[2] |
| Treatment | Avoiding triggers, medications for infection, stopping offending medication, blood transfusions[3] |
| Frequency | 400 million[1] |
| Deaths | 33,000 (2015)[4] |
It is an X-linked recessive disorder that results in defective glucose-6-phosphate dehydrogenase enzyme.[1] Red blood cell breakdown may be triggered by infections, certain medication, stress, or foods such as fava beans.[1][3] Depending on the specific mutation the severity of the condition may vary.[2] Diagnosis is based on symptoms and supported by blood tests and genetic testing.[2]
Avoiding triggers is important.[3] Treatment of acute episodes may include medications for infection, stopping the offending medication, or blood transfusions.[3] Jaundice in newborns may be treated with bili lights.[2] It is recommended that people be tested for G6PDD before certain medications, such as primaquine, are taken.[2]
About 400 million people have the condition globally.[1] It is particularly common in certain parts of Africa, Asia, the Mediterranean, and the Middle East.[1] Males are affected more often than females.[1] In 2015 it is believed to have resulted in 33,000 deaths.[4]
Signs and symptoms
Most individuals with G6PD deficiency are asymptomatic.
Most people who develop symptom are male, due to the X-linked pattern of inheritance, but female carriers can be affected due to unfavorable lyonization, where random inactivation of an X-chromosome in certain cells creates a population of G6PD-deficient red blood cells coexisting with unaffected red blood cells. A female with one affected X chromosome will show the deficiency in approximately half of their red blood cells. However, in rare cases, including double X-deficiency, the ratio can be much more than half, making the individual almost as sensitive as males.
Red blood cell breakdown (also known as hemolysis) in G6PD deficiency can manifest in a number of ways, including the following:
- Prolonged neonatal jaundice, possibly leading to kernicterus (arguably the most serious complication of G6PD deficiency)
- Hemolytic crises in response to:
- Illness (especially infections)
- Certain drugs (see below)
- Certain foods, most notably broad beans, from which the word favism derives
- Certain chemicals
- Diabetic ketoacidosis
- Hemoglobinuria (red or brown urine)
- Very severe crises can cause acute kidney failure
Favism is a hemolytic response to the consumption of fava beans, also known as broad beans. Though all individuals with favism show G6PD deficiency, not all individuals with G6PD deficiency show favism. The condition is known to be more prevalent in infants and children, and G6PD genetic variant can influence chemical sensitivity.[5] Other than this, the specifics of the chemical relationship between favism and G6PD are not well understood.
Cause
Triggers
Carriers of the underlying mutation do not show any symptoms unless their red blood cells are exposed to certain triggers, which can be of four main types:
- Foods (fava beans is the hallmark trigger for G6PD mutation carriers),
- Certain medicines including aspirin, quinine and other antimalarials derived from quinine.
- Moth balls (naphthalene)[6]
- Stress from a bacterial or viral infection.[7]
Drugs
Many substances are potentially harmful to people with G6PD deficiency. Variation in response to these substances makes individual predictions difficult. Antimalarial drugs that can cause acute hemolysis in people with G6PD deficiency include primaquine, pamaquine, chloroquine, and hydroxychloroquine.[8] There is evidence that other antimalarials may also exacerbate G6PD deficiency, but only at higher doses. Sulfonamides (such as sulfanilamide, sulfamethoxazole, and mafenide), thiazolesulfone, methylene blue, and naphthalene should also be avoided by people with G6PD deficiency as they antagonize folate synthesis, as should certain analgesics (such as phenazopyridine and acetanilide) and a few non-sulfa antibiotics (nalidixic acid, nitrofurantoin, isoniazid, dapsone, and furazolidone).[9][10][11] Henna has been known to cause hemolytic crisis in G6PD-deficient infants.[12] Rasburicase is also contraindicated in G6PD deficiency. High dose intravenous vitamin C has also been known to cause haemolysis in G6PD deficiency carriers;[13][14] thus, G6PD deficiency testing is routine before infusion of doses of 25 g or more.
Genetics
Two variants (G6PD A− and G6PD Mediterranean) are the most common in human populations. G6PD A− has an occurrence of 10% of Africans and African-Americans while G6PD Mediterranean is prevalent in the Middle East. The known distribution of the mutated allele is largely limited to people of Mediterranean origins (Spaniards, Italians, Greeks, Armenians, Sephardi Jews and other Semitic peoples).[15] Both variants are believed to stem from a strongly protective effect against Plasmodium falciparum and Plasmodium vivax malaria.[16] It is particularly frequent in the Kurdish Jewish population, wherein approximately 1 in 2 males have the condition and the same rate of females are carriers.[7] It is also common in African American, Saudi Arabian, Sardinian males, some African populations, and Asian groups.[7]
All mutations that cause G6PD deficiency are found on the long arm of the X chromosome, on band Xq28. The G6PD gene spans some 18.5 kilobases.[10] The following variants and mutations are well-known and described:
| Descriptive mutations | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mutation | Gene | Protein | |||||||
| Designation | Short name | Isoform G6PD-Protein |
OMIM-Code | Type | Subtype | Position | Position | Structure change | Function change |
| G6PD-A(+) | Gd-A(+) | G6PD A | +305900.0001 | Polymorphism nucleotide | A→G | 376 (Exon 5) |
126 | Asparagine→Aspartic acid (ASN126ASP) | No enzyme defect (variant) |
| G6PD-A(-) | Gd-A(-) | G6PD A | +305900.0002 | Substitution nucleotide | G→A | 376 (Exon 5) and 202 |
68 and 126 |
Valine→Methionine (VAL68MET) Asparagine→Aspartic acid (ASN126ASP) |
|
| G6PD-Mediterranean | Gd-Med | G6PD B | +305900.0006 | Substitution nucleotide | C→T | 563 (Exon 6) |
188 | Serine→Phenylalanine (SER188PHE) | Class II |
| G6PD-Canton | Gd-Canton | G6PD B | +305900.0021 | Substitution nucleotide | G→T | 1376 | 459 | Arginine→Leucine (ARG459LEU) | Class II |
| G6PD-Chatham | Gd-Chatham | G6PD | +305900.0003 | Substitution nucleotide | G→A | 1003 | 335 | Alanine→Threonine (ALA335THR) | Class II |
| G6PD-Cosenza | Gd-Cosenza | G6PD B | +305900.0059 | Substitution nucleotide | G→C | 1376 | 459 | Arginine→Proline (ARG459PRO) | G6PD-activity <10%, thus high portion of patients. |
| G6PD-Mahidol | Gd-Mahidol | G6PD | +305900.0005 | Substitution nucleotide | G→A | 487 (Exon 6) |
163 | Glycine→Serine (GLY163SER) | Class III |
| G6PD-Orissa | Gd-Orissa | G6PD | +305900.0047 | Substitution nucleotide | C→G | 131 | 44 | Alanine→Glycine (ALA44GLY) | NADP-binding place affected. Higher stability than other variants. |
| G6PD-Asahi | Gd-Asahi | G6PD A- | +305900.0054 | Substitution nucleotide (several) | A→G ± G→A |
376 (Exon 5) 202 |
126 68 |
Asparagine→Aspartic acid (ASN126ASP) Valine→Methionine (VAL68MET) |
Class III. |
Pathophysiology
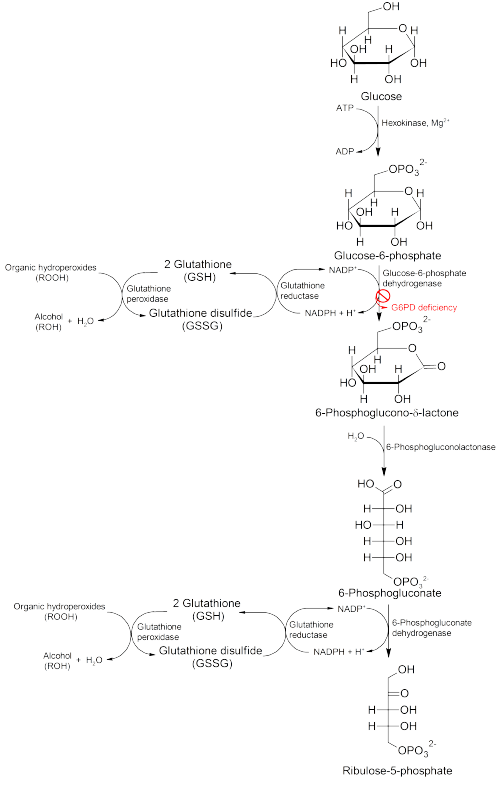
Glucose-6-phosphate dehydrogenase (G6PD) is an enzyme in the pentose phosphate pathway (see image, also known as the HMP shunt pathway). G6PD converts glucose-6-phosphate into 6-phosphoglucono-δ-lactone and is the rate-limiting enzyme of this metabolic pathway that supplies reducing energy to cells by maintaining the level of the reduced form of the co-enzyme nicotinamide adenine dinucleotide phosphate (NADPH). The NADPH in turn maintains the supply of reduced glutathione in the cells that is used to mop up free radicals that cause oxidative damage.
The G6PD / NADPH pathway is the only source of reduced glutathione in red blood cells (erythrocytes). The role of red cells as oxygen carriers puts them at substantial risk of damage from oxidizing free radicals except for the protective effect of G6PD/NADPH/glutathione.
People with G6PD deficiency are therefore at risk of hemolytic anemia in states of oxidative stress. Oxidative stress can result from infection and from chemical exposure to medication and certain foods. Broad beans, e.g., fava beans, contain high levels of vicine, divicine, convicine and isouramil, all of which create oxidants.[17]
When all remaining reduced glutathione is consumed, enzymes and other proteins (including hemoglobin) are subsequently damaged by the oxidants, leading to cross-bonding and protein deposition in the red cell membranes. Damaged red cells are phagocytosed and sequestered (taken out of circulation) in the spleen. The hemoglobin is metabolized to bilirubin (causing jaundice at high concentrations). The red cells rarely disintegrate in the circulation, so hemoglobin is rarely excreted directly by the kidney, but this can occur in severe cases, causing acute kidney injury.
Deficiency of G6PD in the alternative pathway causes the buildup of glucose and thus there is an increase of advanced glycation endproducts (AGE). The deficiency also reduces the amount of NADPH, which is required for the formation of nitric oxide (NO). The high prevalence of diabetes mellitus type 2 and hypertension in Afro-Caribbeans in the West could be directly related to the incidence of G6PD deficiency in those populations.[18]
Although female carriers can have a mild form of G6PD deficiency (dependent on the degree of inactivation of the unaffected X chromosome – see lyonization), homozygous females have been described; in these females there is co-incidence of a rare immune disorder termed chronic granulomatous disease (CGD).
Diagnosis
The diagnosis is generally suspected when patients from certain ethnic groups (see epidemiology) develop anemia, jaundice and symptoms of hemolysis after challenges from any of the above causes, especially when there is a positive family history.
Generally, tests will include:
- Complete blood count and reticulocyte count; in active G6PD deficiency, Heinz bodies can be seen in red blood cells on a blood film;
- Liver enzymes (to exclude other causes of jaundice);
- Lactate dehydrogenase (elevated in hemolysis and a marker of hemolytic severity)
- Haptoglobin (decreased in hemolysis);
- A "direct antiglobulin test" (Coombs' test) – this should be negative, as hemolysis in G6PD is not immune-mediated;
When there are sufficient grounds to suspect G6PD, a direct test for G6PD is the "Beutler fluorescent spot test", which has largely replaced an older test (the Motulsky dye-decolouration test). Other possibilities are direct DNA testing and/or sequencing of the G6PD gene.
The Beutler fluorescent spot test is a rapid and inexpensive test that visually identifies NADPH produced by G6PD under ultraviolet light. When the blood spot does not fluoresce, the test is positive; it can be falsely negative in patients who are actively hemolysing. It can therefore only be done 2–3 weeks after a hemolytic episode.
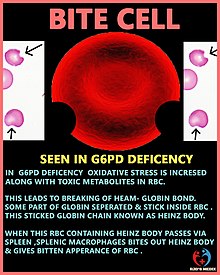
When a macrophage in the spleen identifies a RBC with a Heinz body, it removes the precipitate and a small piece of the membrane, leading to characteristic "bite cells". However, if a large number of Heinz bodies are produced, as in the case of G6PD deficiency, some Heinz bodies will nonetheless be visible when viewing RBCs that have been stained with crystal violet. This easy and inexpensive test can lead to an initial presumption of G6PD deficiency, which can be confirmed with the other tests.
Classification
The World Health Organization classifies G6PD genetic variants into five classes, the first three of which are deficiency states.[19]
- Class I: Severe deficiency (<10% activity) with chronic (nonspherocytic) hemolytic anemia
- Class II: Severe deficiency (<10% activity), with intermittent hemolysis
- Class III: Moderate deficiency (10-60% activity), hemolysis with stressors only
- Class IV: Non-deficient variant, no clinical sequelae
- Class V: Increased enzyme activity, no clinical sequelae
Differential diagnosis
6-phosphogluconate dehydrogenase (6PGD) deficiency has similar symptoms and is often mistaken for G6PD deficiency, as the affected enzyme is within the same pathway, however these diseases are not linked and can be found within the same person.
Treatment
The most important measure is prevention – avoidance of the drugs and foods that cause hemolysis. Vaccination against some common pathogens (e.g. hepatitis A and hepatitis B) may prevent infection-induced attacks.[20]
In the acute phase of hemolysis, blood transfusions might be necessary, or even dialysis in acute kidney failure. Blood transfusion is an important symptomatic measure, as the transfused red cells are generally not G6PD deficient and will live a normal lifespan in the recipient's circulation. Those affected should avoid drugs such as aspirin.
Some patients may benefit from removal of the spleen (splenectomy),[21] as this is an important site of red cell destruction. Folic acid should be used in any disorder featuring a high red cell turnover. Although vitamin E and selenium have antioxidant properties, their use does not decrease the severity of G6PD deficiency.
Prognosis
G6PD-deficient individuals do not appear to acquire any illnesses more frequently than other people, and may have less risk than other people for acquiring ischemic heart disease and cerebrovascular disease.[22]
Epidemiology
G6PD deficiency is the second most common human enzyme defect after ALDH2 deficiency, being present in more than 400 million people worldwide.[23] G6PD deficiency resulted in 4,100 deaths in 2013 and 3,400 deaths in 1990.[24] African, Middle Eastern and South Asian people are affected the most, including those who have these ancestries.[25] [26] A side effect of this disease is that it confers protection against malaria,[27] in particular the form of malaria caused by Plasmodium falciparum, the most deadly form of malaria. A similar relationship exists between malaria and sickle-cell disease. One theory to explain this is that cells infected with the Plasmodium parasite are cleared more rapidly by the spleen. This phenomenon might give G6PD deficiency carriers an evolutionary advantage by increasing their fitness in malarial endemic environments. In vitro studies have shown that the Plasmodium falciparum is very sensitive to oxidative damage. This is the basis for another theory, that is that the genetic defect confers resistance due to the fact that the G6PD-deficient host has a higher level of oxidative agents that, while generally tolerable by the host, are deadly to the parasite.[28]
History
The modern understanding of the condition began with the analysis of patients who exhibited sensitivity to primaquine.[29] The discovery of G6PD deficiency relied heavily upon the testing of prisoner volunteers at Illinois State Penitentiary, a type of study which today is considered unethical and cannot be performed. When some prisoners were given the drug primaquine, some developed hemolytic anemia but others did not. In spite of these results, the US military administered the drug widely during the Korean War to prevent the relapsing infection caused by Plasmodium vivax hypnozoites. Numerous cases of hemolytic anemia were observed in US soldiers of North African and Mediterranean descent.[30]
After studying the mechanism through Cr51 testing, it was conclusively shown that the hemolytic effect of primaquine was due to an intrinsic defect of erythrocytes.[31]
Society and culture
In both legend and mythology, favism has been known since antiquity. The priests of various Greco-Roman era cults were forbidden to eat or even mention beans, and Pythagoras had a strict rule that to join the society of the Pythagoreans one had to swear off beans.[32] This ban was supposedly because beans resembled male genitalia, but it is possible that this was because of a belief that beans and humans were created from the same material.[33]
References
- "Glucose-6-phosphate dehydrogenase deficiency". Genetics Home Reference. 6 December 2017. Retrieved 10 December 2017.
- "Glucose-6-Phosphate Dehydrogenase Deficiency". NORD (National Organization for Rare Disorders). 2017. Retrieved 11 December 2017.
- "Glucose-6-phosphate dehydrogenase deficiency". Genetic and Rare Diseases Information Center (GARD). 2017. Retrieved 10 December 2017.
- GBD 2015 Mortality and Causes of Death, Collaborators. (8 October 2016). "Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015". Lancet. 388 (10053): 1459–1544. doi:10.1016/s0140-6736(16)31012-1. PMC 5388903. PMID 27733281.
- Luzzatto, L. "GLUCOSE-6-PHOSPHATE DEHYDROGENASE DEFICIENCY." Advanced Medicine-Twelve: Proceedings of a Conference Held at the Royal College of Physicians of London, 11–14 February 1985. Vol. 21. Churchill Livingstone, 1986.
- "Triggers of G6PD crisis" (PDF). Sydney Local Health District.
- Glucose-6-Phosphate Dehydogenase Deficiency (G6PD) on The Jewish Genetic Disease Consortium (JGDC) website .Archived 1 July 2017 at the Wayback Machine
- Jones, & Bartlett; Publishers, Jones and Bartlett (2010). 2010 Nurse's Drug Handbook. Jones & Bartlett Learning. p. 497. ISBN 978-0-7637-7900-9.
- Frank JE (October 2005). "Diagnosis and management of G6PD deficiency". Am Fam Physician. 72 (7): 1277–82. PMID 16225031.
- Warrell, David A.; Timothy M. Cox; John D. Firth; Edward J. Benz (2005). Oxford Textbook of Medicine, Volume Three. Oxford University Press. pp. 720–725. ISBN 978-0-19-857013-4.
- A comprehensive list of drugs and chemicals that are potentially harmful in G6PD deficiency can be found in Beutler E (December 1994). "G6PD deficiency". Blood. 84 (11): 3613–36. doi:10.1182/blood.V84.11.3613.bloodjournal84113613. PMID 7949118..
- Raupp P, Hassan JA, Varughese M, Kristiansson B (2001). "Henna causes life threatening haemolysis in glucose-6-phosphate dehydrogenase deficiency". Arch. Dis. Child. 85 (5): 411–2. doi:10.1136/adc.85.5.411. PMC 1718961. PMID 11668106.
- Rees, DC; Kelsey, H; Richards, JD (27 March 1993). "Acute haemolysis induced by high dose ascorbic acid in glucose-6-phosphate dehydrogenase deficiency". BMJ (Clinical Research Ed.). 306 (6881): 841–2. doi:10.1136/bmj.306.6881.841. PMC 1677333. PMID 8490379.
- Mehta, JB; Singhal, SB; Mehta, BC (13 October 1990). "Ascorbic-acid-induced haemolysis in G-6-PD deficiency". Lancet. 336 (8720): 944. doi:10.1016/0140-6736(90)92317-b. PMID 1976956.
- "Favism | genetic disorder".
- Kumar, Vinay; Abbas, Abul K.; Fausto, Nelson; Aster, Jon (2009-05-28). Robbins and Cotran Pathologic Basis of Disease, Professional Edition: Expert Consult - Online (Robbins Pathology) (Kindle Locations 33351-33354). Elsevier Health. Kindle Edition.
- Chevion, Mordechai; Navok, Tikva; Glaser, Gad; Mager, Jacob (1982-10-01). "The Chemistry of Favism-Inducing Compounds". European Journal of Biochemistry. 127 (2): 405–409. doi:10.1111/j.1432-1033.1982.tb06886.x. ISSN 1432-1033.
- Gaskin RS, Estwick D, Peddi R (2001). "G6PD deficiency: its role in the high prevalence of hypertension and diabetes mellitus". Ethnicity & Disease. 11 (4): 749–54. PMID 11763298.
- WHO Working Group (1989). "Glucose-6-phosphate dehydrogenase deficiency". Bulletin of the World Health Organization. 67 (6): 601–11. PMC 2491315. PMID 2633878.
- Monga A, Makkar RP, Arora A, Mukhopadhyay S, Gupta AK (July 2003). "Case report: Acute hepatitis E infection with coexistent glucose-6-phosphate dehydrogenase deficiency". Can J Infect Dis. 14 (4): 230–1. doi:10.1155/2003/913679. PMC 2094938. PMID 18159462.
- Hamilton JW, Jones FG, McMullin MF (August 2004). "Glucose-6-phosphate dehydrogenase Guadalajara – a case of chronic non-spherocytic haemolytic anaemia responding to splenectomy and the role of splenectomy in this disorder". Hematology. 9 (4): 307–9. doi:10.1080/10245330410001714211. PMID 15621740.
- thefreedictionary.com > glucose-6-phosphate dehydrogenase deficiency citing: Gale Encyclopedia of Medicine. Copyright 2008
- Cappellini MD, Fiorelli G (January 2008). "Glucose-6-phosphate dehydrogenase deficiency". Lancet. 371 (9606): 64–74. doi:10.1016/S0140-6736(08)60073-2. PMID 18177777.
- GBD 2013 Mortality and Causes of Death, Collaborators (17 December 2014). "Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: a systematic analysis for the Global Burden of Disease Study 2013". Lancet. 385 (9963): 117–71. doi:10.1016/S0140-6736(14)61682-2. PMC 4340604. PMID 25530442.
- Gelabert, Pere; Olalde, Iñigo; de-Dios, Toni; Civit, Sergi; Lalueza-Fox, Carles (2017). "Malaria was a weak selective force in ancient Europeans". Scientific Reports. 7 (1): 1377. Bibcode:2017NatSR...7.1377G. doi:10.1038/s41598-017-01534-5. ISSN 2045-2322. PMC 5431260. PMID 28469196.
- G-6-PD FAQ section
- Mehta A, Mason PJ, Vulliamy TJ (2000). "Glucose-6-phosphate dehydrogenase deficiency". Best Practice & Research Clinical Haematology. 13 (1): 21–38. doi:10.1053/beha.1999.0055. PMC 2398001. PMID 10916676.
- Nelson, David L.; Cox, Michael M. (13 February 2013). Lehninger Principles of Biochemistry (6th ed.). Basingstoke, England: Macmillan Higher Education. p. 576. ISBN 978-1-4641-0962-1.
- Alving AS, Carson PE, Flanagan CL, Ickes CE (September 1956). "Enzymatic deficiency in primaquine-sensitive erythrocytes". Science. 124 (3220): 484–5. Bibcode:1956Sci...124..484C. doi:10.1126/science.124.3220.484-a. PMID 13360274.
- Baird K (2015). "Origins and implications of neglect of G6PD deficiency and primaquine toxicity in Plasmodium vivax malaria". Pathog Glob Health. 109 (3): 93–106. doi:10.1179/2047773215Y.0000000016. PMC 4455359. PMID 25943156.
- Beutler E (January 2008). "Glucose-6-phosphate dehydrogenase deficiency: a historical perspective". Blood. 111 (1): 16–24. doi:10.1182/blood-2007-04-077412. PMID 18156501.
- Simoons, F.J. (1996-08-30). "8". Plants of Life, Plants of Death. University of Wisconsin Press. p. 216. ISBN 978-0299159047.
- Rendall, Steven; Riedweg, Christoph (2005). Pythagoras: his life, teaching, and influence. Ithaca, N.Y: Cornell University Press. ISBN 978-0-8014-4240-7.
External links
| Classification | |
|---|---|
| External resources |