Menkes disease
Menkes disease (MNK), also known as Menkes syndrome,[1][2] is an X-linked recessive disorder caused by mutations in genes coding for the copper-transport protein ATP7A,[3] leading to copper deficiency.[4][5] Characteristic findings include kinky hair, growth failure, and nervous system deterioration. Like all X-linked recessive conditions, Menkes disease is more common in males than in females. The disorder was first described by John Hans Menkes in 1962.[6]
| Menkes disease | |
|---|---|
| Other names | Trichopoliodystrophy, copper transport disease, steely hair disease, kinky hair disease |
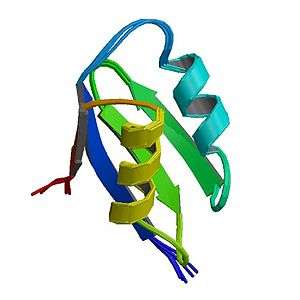 | |
| ATP7A | |
| Specialty | Endocrinology |
Onset occurs during infancy, with incidence of about 1 in 100,000 to 250,000 newborns; affected infants often do not live past the age of three years, though there are rare cases in which less severe symptoms emerge later in childhood.[7]
Signs and symptoms
Affected infants may be born prematurely. Signs and symptoms appear during infancy, typically after a two- to three-month period of normal or slightly slowed development that is followed by a loss of early developmental skills and subsequent developmental delay. Patients exhibit hypotonia (weak muscle tone), failure to thrive, hypothermia (subnormal body temperature), sagging facial features, seizures, and metaphyseal widening. Hair appears strikingly peculiar: kinky, colorless or silvery, and brittle. There can be extensive neurodegeneration in the gray matter of the brain.[8] Arteries in the brain can also be twisted with frayed and split inner walls. This can lead to rupture or blockage of the arteries. Weakened bones (osteoporosis) may result in fractures.
Occipital horn syndrome (sometimes called X-linked cutis laxa or Ehlers-Danlos type 9[9]) is a mild form of Menkes syndrome that begins in early to middle childhood. It is characterized by calcium deposits in a bone at the base of the skull (occipital bone), coarse hair, and loose skin and joints.
Cause
Mutations in the ATP7A gene, located on chromosome Xq21.1,[10] leads to Menkes syndrome.[11] This condition is inherited in an X-linked recessive pattern.[12] About 30% of MNK cases are due to new mutations and 70% are inherited, almost always from the mother.[7] Even though the disease is more common in males, females can still be a carrier of the disease. As the result of a mutation in the ATP7A gene, copper is poorly distributed to cells in the body. Copper accumulates in some tissues, such as the small intestine and kidneys, while the brain and other tissues have unusually low levels. The decreased supply of copper can reduce the activity of numerous copper-containing enzymes that are necessary for the structure and function of bone, skin, hair, blood vessels and the nervous system such as lysyl oxidase.[13] As with other X-linked disorders, female children of a carrier mother have an even chance of carrying the disorder, but are normally well; male children have an even chance of having the disorder or of being free from it. A genetic counselor may have useful advice.
Mechanism
The ATP7A gene encodes a transmembrane protein that transport copper across the cell membranes. It is found throughout the body, except for the liver. In the small intestines, the ATP7A protein helps control the absorption of copper from food. In other cells, the protein travels between the Golgi apparatus and the cell membrane to maintain copper concentrations in the cell. The protein is normally found in the Golgi apparatus, which is important for modifying proteins, including enzymes. In the Golgi apparatus, ATP7A protein provides copper to certain enzymes that are critical for the structure and function of bone, skin, hair, blood vessels, and the nervous system.[14] One of the enzymes, lysyl oxidase, requires copper for proper function. This enzyme cross-links tropocollagen into strong collagen fibrils. The defective collagen contributes to many of the aforementioned connective tissue manifestations of this disease.
If copper levels become excessive, the protein will travel to the cell membrane and eliminate excess copper from the cell. Mutations in the ATP7A gene such as deletions and insertions lead to parts of the gene being deleted, resulting in a shortened ATP7A protein. This prevents the production of a functional ATP7A protein, leading to the impaired absorption of copper from food and copper will not be supplied to certain enzymes.
Diagnosis
Menkes syndrome can be diagnosed by blood tests of the copper and ceruloplasmin levels, skin biopsy, and optical microscopic examination of the hair to view characteristic Menkes abnormalities. X-rays of the skull and skeleton are conducted to look for abnormalities in bone formation.[7] Urine homovanillic acid/vanillylmandelic acid ratio has been proposed as a screening tool to support earlier detection.[15][16] Since 70% of MNK cases are inherited, genetic testing of the mother can be performed to search for a mutation in the ATP7A gene.
Treatment
There is no cure for Menkes disease. Early treatment with injections of copper supplements (acetate or glycinate) may be of some slight benefit. Among 12 newborns who were diagnosed with MNK, 92% were alive at age 4.6.[17] Other treatment is symptomatic and supportive. Treatments to help relieve some of the symptoms includes pain medication, anti-seizure medication, feeding tube when necessary, and physical and occupational therapy.[18] The earlier treatment is given, the better the prognosis.[19]
Epidemiology
One European study reported a rate of 1 in 254,000;[20] a Japanese study reported a rate of 1 in 357,143.[21] No correlation with other inherited characteristics, or with ethnic origin, is known.
See also
References
- Online Mendelian Inheritance in Man (OMIM): 309400
- James, William; Berger, Timothy; Elston, Dirk (2005). Andrews Diseases of the Skin: Clinical Dermatology (10th ed.). Saunders. p. 765. ISBN 978-0-7216-2921-6.
- "Menkes syndrome" at Dorland's Medical Dictionary
- Vest, Katherine E.; Hashemi, Hayaa F.; Cobine, Paul A. (2013). "Chapter 13 The Copper Metallome in Eukaryotic Cells". In Banci, Lucia (ed.). Metallomics and the Cell. Metal Ions in Life Sciences. 12. Springer. doi:10.1007/978-94-007-5561-10_12 (inactive 2020-01-22). ISBN 978-94-007-5560-4. electronic-book ISBN 978-94-007-5561-1 ISSN 1559-0836 electronic-ISSN 1868-0402
- de Bie P, Muller P, Wijmenga C, Klomp LW (Nov 2007). "Molecular pathogenesis of Wilson and Menkes disease: correlation of mutations with molecular defects and disease phenotypes". J. Med. Genet. 44 (11): 673–688. doi:10.1136/jmg.2007.052746. PMC 2752173. PMID 17717039.
- Menkes JH, Alter M, Steigleder GK, Weakley DR, Sung JH (1962). "A sex-linked recessive disorder with retardation of growth, peculiar hair, and focal cerebral and cerebellar degeneration". Pediatrics. 29: 764–779. PMID 14472668.
- "Research Overview". themenkesfoundation.org. Archived from the original on 2017-02-12. Retrieved 2015-12-10.
- Barnes N, Tsivkovskii R, Tsivkovskaia N, Lutsenko S (2005). "The copper-transporting ATPases, Menkes and Wilson disease proteins, have distinct roles in adult and developing cerebellum". J Biol Chem. 280 (10): 9640–5. doi:10.1074/jbc.M413840200. PMID 15634671.
- Menkes Disease at eMedicine
- Online Mendelian Inheritance in Man (OMIM): 300011
- Voskoboinik I, Camakaris J (2002). "Menkes copper-translocating P-type ATPase (ATPTA): biochemical and cell biology properties, and role in Menkes disease". J Bioenerg Biomembr. 34 (5): 363–71. doi:10.1023/A:1021250003104. PMID 12539963.
- Kim BE, Smith K, Meagher CK, Petris MJ (November 2002). "A conditional mutation affecting localization of the Menkes disease copper ATPase. Suppression by copper supplementation". J. Biol. Chem. 277 (46): 44079–84. doi:10.1074/jbc.M208737200. PMID 12221109.
- Scheiber, Ivo; Dringen, Ralf; Mercer, Julian F. B. (2013). "Chapter 11. Copper: Effects of Deficiency and Overload". In Astrid Sigel, Helmut Sigel and Roland K. O. Sigel (ed.). Interrelations between Essential Metal Ions and Human Diseases. Metal Ions in Life Sciences. 13. Springer. pp. 359–387. doi:10.1007/978-94-007-7500-8_11. ISBN 978-94-007-7499-5. PMID 24470097.
- "ATP7A gene". Genetics Home Reference. 2015-12-07. Retrieved 2015-12-10.
- Matsuo M, Tasaki R, Kodama H, Hamasaki Y (2005). "Screening for Menkes disease using the urine HVA/VMA ratio". J. Inherit. Metab. Dis. 28 (1): 89–93. doi:10.1007/s10545-005-5083-6. PMID 15702409.
- "Role of optic microscopy for early diagnosis of Menkes disease". ResearchGate. Retrieved 2015-12-10.
- Kaler SG, Holmes CS, Goldstein DS (February 2008). "Neonatal diagnosis and treatment of Menkes disease". N. Engl. J. Med. 358 (6): 605–14. doi:10.1056/NEJMoa070613. PMC 3477514. PMID 18256395.
- Kaler, Stephen G.; Holmes, Courtney S.; Goldstein, David S.; Tang, Jingrong; Godwin, Sarah C.; Donsante, Anthony; Liew, Clarissa J.; Sato, Susumu; Patronas, Nicholas (2008-02-07). "Neonatal Diagnosis and Treatment of Menkes Disease". New England Journal of Medicine. 358 (6): 605–614. doi:10.1056/NEJMoa070613. ISSN 0028-4793. PMC 3477514. PMID 18256395.
- "Research Overview". themenkesfoundation.org. Archived from the original on 2017-02-12. Retrieved 2018-04-08.
- Tønnesen T, Kleijer WJ, Horn N (February 1991). "Incidence of Menkes disease". Hum. Genet. 86 (4): 408–10. doi:10.1007/BF00201846. PMID 1999344.
- Gu YH, Kodama H, Shiga K, Nakata S, Yanagawa Y, Ozawa H (2005). "A survey of Japanese patients with Menkes disease from 1990 to 2003: incidence and early signs before typical symptomatic onset, pointing the way to earlier diagnosis". J. Inherit. Metab. Dis. 28 (4): 473–8. doi:10.1007/s10545-005-0473-3. PMID 15902550.
External links
| Classification | |
|---|---|
| External resources |
| Wikimedia Commons has media related to Menkes disease. |