Glycogen storage disease type VI
Glycogen storage disease type VI (GSD VI) is a type of glycogen storage disease caused by a deficiency in liver glycogen phosphorylase or other components of the associated phosphorylase cascade system.[2] It is also known as "Hers' disease", after Henri G. Hers, who characterized it in 1959.[3] The scope of GSD VI now also includes glycogen storage disease type VIII,[2] IX[2] (caused by phosphorylase b kinase deficiency) and X[2] (deficiency protein kinase A).
| Glycogen storage disease type VI | |
|---|---|
| Other names | Glycogen storage disease due to liver glycogen phosphorylase deficiency[1] |
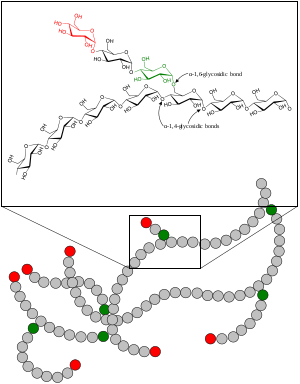 | |
| Glycogen | |
| Specialty | Endocrinology |
The incidence of GSD VI is approximately 1 case per 65,000–85,000 births,[2] representing approximately 30% all cases of glycogen storage disease.
Signs/symptoms
Patients generally have a benign course, and typically present with hepatomegaly and growth retardation early in childhood. Mild hypoglycemia, hyperlipidemia, and hyperketosis may occur. Lactic acid and uric acid levels may be normal. However, lactic acidosis may occur during fasting.[4]
Diagnosis
Treatment
See also
References
- RESERVED, INSERM US14-- ALL RIGHTS. "Orphanet: Glycogen storage disease due to liver glycogen phosphorylase deficiency". www.orpha.net. Retrieved 11 April 2019.
- Glycogen-Storage Disease Type VI at eMedicine
- Hers HG (1959). "[Enzymatic studies of hepatic fragments; application to the classification of glycogenoses.]". Rev Int Hepatol (in French). 9 (1): 35–55. PMID 13646331.
- "Glycogen storage disease type VI".
Further reading
External links

| Classification | |
|---|---|
| External resources |