6-phosphogluconate dehydrogenase deficiency
6-Phosphogluconate dehydrogenase deficiency (6PGD deficiency), or partial deficiency, is an autosomal hereditary disease characterized by abnormally low levels of 6-Phosphogluconate dehydrogenase (6PGD), a metabolic enzyme involved in the Pentose phosphate pathway. It is very important in the metabolism of red blood cells (erythrocytes). 6PDG deficiency affects less than 1% of the population, and studies suggest that there may be race variant involved in many of the reported cases. Although it is similar, 6PDG deficiency is not linked to glucose-6-phosphate dehydrogenase deficiency, as they are located on different chromosomes. However, a few people have had both of these metabolic diseases.
| 6-phosphogluconate dehydrogenase deficiency | |
|---|---|
 | |
| Crystallographic structure of sheep 6-phosphogluconate dehydrogenase complexed with adenosine 2'-monophosphate.[1] | |
| Specialty | Hematology |
| 6-phosphogluconate dehydrogenase | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Identifiers | |||||||||
| Symbol | 6PGD | ||||||||
| Pfam | PF00393 | ||||||||
| Pfam clan | CL0106 | ||||||||
| InterPro | IPR006114 | ||||||||
| PROSITE | PDOC00390 | ||||||||
| SCOPe | 2pgd / SUPFAM | ||||||||
| |||||||||
Signs and symptoms
6-Phosphogluconate dehydrogenase deficiency can be asymptomatic in many patients who are carriers. Female carriers have been found to be at a higher frequency experiencing symptoms. Enzyme activity was shown to be reduced by 35–65% depending on the severity of the deficiency.
Abnormal red blood cell breakdown (hemolysis) in 6PGD deficiency can be symptomatic in a number of ways, including the following:
- Neonatal jaundice,
- Possibility of leading to kernicterus due to a hyperbilirubinemia
- Hemolytic crises in response to:
- Illness and infections
- Certain drugs
- Certain foods
- Certain chemicals
- In extreme cases, kidney failure
Genetics
6PGD deficiency is a recessive hereditary disorder located on the P arm of chromosome 1. It is an autosomal disease, not associated with the sex chromosomes and can affect both sexes. The lack of synthesis of a specific protein on chromosome 1 has reduced a subject suffering from 6PGD deficiency from producing adequate amounts of the 6-Phosphogluconate dehydrogenase enzyme. Transfer of the disease can be passed from a parent, even when the parent is asymptomatic.
Pathophysiology
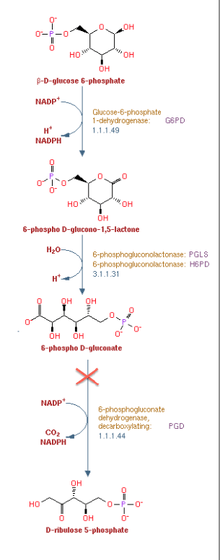
Reaction mechanism
6-Phosphogluconate dehydrogenase (6PGD) is an enzyme in the pentose phosphate pathway (see image). 6PGD catalyzes the reaction of 6-phosphogluconate to an unstable form of 3-keto-6-phosphogluconate, and yields a co-enzyme nicotinamide adenine dinucleotide phosphate (NADPH) as a byproduct. NADPH supplies reducing power to cells. The reaction is the second NADPH releasing reaction in the pentose phosphate pathway, the first being catalyzed by glucose-6-phosphate dehydrogenase. 3-keto-6-phosphogluconate then rapidly (in an irreversible reaction) decarboxylates to CO2 and ribulose-5-phosphate, which is the precursor to many vital metabolic processes.
Importance of NADPH
The NADPH pathway (both 6PGD and G6PD reactions) is the only source of reductant to reduce glutathione in red blood cells. The role of erythrocytes as oxygen carriers puts them at risk of being damaged by oxidizing free radicals. The reduction of glutathione acts as an antioxidant and prevents damage from reactive oxygen species.
Oxidative stress
People suffering from 6PGD and/or G6PD deficiencies are therefore at risk of hemolytic anemia in states of oxidative stress. Oxidative stress can result from infection and from chemical exposure to medication and certain foods. Broad beans, e.g., fava beans, contain high levels of vicine, divicine, convicine and isouramil, all of which are oxidants.
When all remaining reduced glutathione is consumed, enzymes and other proteins, such as hemoglobin are subsequently damaged by the free radicals, leading to electrolyte imbalance, cross-bonding and protein deposition in the red cell membranes. Damaged red cells are phagocytosed and sequestered (taken out of circulation) in the spleen. The hemoglobin is metabolized to bilirubin (causing jaundice). The red blood cells rarely disintegrate in the circulation, so hemoglobin is rarely excreted directly by the kidney, but this can occur in severe cases, causing acute kidney injury.
Diagnosis
Treatment
Prevention
The most important measure taken for treatment of 6-phosphoglucanate dehydrogenase is prevention. Avoidance of chemical exposures to drugs and foods that have the potential to cause hemolysis. Although some foods and supplements have antioxidant properties, their use does not decrease the severity of G6PD deficiency.
Diagnosis is difficult during haemolytic episodes since reticulocytes have increased levels of enzymes and may get erroneously normal result. wait until steady state (about 6weeks after episodes of haemolysis.
G6PD assay to confirm diagnosis G6PD spectrophotometry to detect level of activity
Vaccinations against some common pathogens (e.g. hepatitis A and hepatitis B) may prevent infection-induced attacks.[2]
Blood transfusion
In the acute phase of hemolysis, blood transfusions might be necessary, or even dialysis in acute kidney injury. Blood transfusion is an important symptomatic measure, as the transfused red cells are generally not 6PGD deficient and will live a normal lifespan in the recipient's circulation.
References
- PDB: 1PGQ; Adams MJ, Ellis GH, Gover S, Naylor CE, Phillips C (July 1994). "Crystallographic study of coenzyme, coenzyme analogue and substrate binding in 6-phosphogluconate dehydrogenase: implications for NADP specificity and the enzyme mechanism". Structure. 2 (7): 651–68. doi:10.1016/s0969-2126(00)00066-6. PMID 7922042.
- Monga A, Makkar RP, Arora A, Mukhopadhyay S, Gupta AK (July 2003). "Case report: Acute hepatitis E infection with coexistent glucose-6-phosphate dehydrogenase deficiency". Can J Infect Dis. 14 (4): 230–1. doi:10.1155/2003/913679. PMC 2094938. PMID 18159462.