Distillation
Distillation is the process of separating the components or substances from a liquid mixture by using selective boiling and condensation. Distillation may result in essentially complete separation (nearly pure components), or it may be a partial separation that increases the concentration of selected components in the mixture. In either case, the process exploits differences in the relative volatility of the mixture's components. In industrial chemistry, distillation is a unit operation of practically universal importance, but it is a physical separation process, not a chemical reaction.
Distillation has many applications. For example:
- The distillation of fermented products produces distilled beverages with a high alcohol content, or separates other fermentation products of commercial value.
- Distillation is an effective and traditional method of desalination.
- In the petroleum industry, oil stabilization is a form of partial distillation that reduces the vapor pressure of crude oil, thereby making it safe for storage and transport as well as reducing the atmospheric emissions of volatile hydrocarbons. In midstream operations at oil refineries, fractional distillation is a major class of operation for transforming crude oil into fuels and chemical feed stocks.[2][3][4]
- Cryogenic distillation leads to the separation of air into its components – notably oxygen, nitrogen, and argon – for industrial use.
- In the chemical industry, large amounts of crude liquid products of chemical synthesis are distilled to separate them, either from other products, from impurities, or from unreacted starting materials.
An installation used for distillation, especially of distilled beverages, is a distillery. The distillation equipment itself is a still.
History
Early evidence of distillation was found on Akkadian tablets dated c. 1200 BC describing perfumery operations. The tablets provided textual evidence that an early primitive form of distillation was known to the Babylonians of ancient Mesopotamia.[8] Early evidence of distillation was also found related to alchemists working in Alexandria in Roman Egypt in the 1st century.[9]
Distilled water has been in use since at least c. 200, when Alexander of Aphrodisias described the process.[10][11] Work on distilling other liquids continued in early Byzantine Egypt under Zosimus of Panopolis in the 3rd century. Distillation was practiced in the ancient Indian subcontinent, which is evident from baked clay retorts and receivers found at Taxila, Shaikhan Dheri, and Charsadda in modern Pakistan, dating to the early centuries of the Common Era.[12][13][14] These "Gandhara stills" were only capable of producing very weak liquor, as there was no efficient means of collecting the vapors at low heat.[15]
Distillation in China may have begun during the Eastern Han dynasty (1st–2nd centuries), but the distillation of beverages began in the Jin (12th–13th centuries) and Southern Song (10th–13th centuries) dynasties, according to archaeological evidence.[16]
Clear evidence of the distillation of alcohol comes from the Arab chemist Al-Kindi in 9th-century Iraq.,[17][18][19][17][15] where it was described by the School of Salerno in the 12th century.[9][20] Fractional distillation was developed by Tadeo Alderotti in the 13th century.[21] A still was found in an archaeological site in Qinglong, Hebei province, in China, dating back to the 12th century. Distilled beverages were common during the Yuan dynasty (13th–14th centuries).[16]
In 1500, German alchemist Hieronymus Braunschweig published Liber de arte destillandi (The Book of the Art of Distillation),[22] the first book solely dedicated to the subject of distillation, followed in 1512 by a much expanded version. In 1651, John French published The Art of Distillation,[23] the first major English compendium on the practice, but it has been claimed[24] that much of it derives from Braunschweig's work. This includes diagrams with people in them showing the industrial rather than bench scale of the operation.


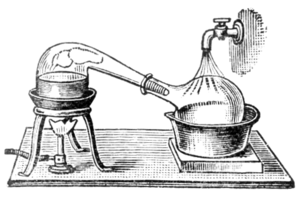


As alchemy evolved into the science of chemistry, vessels called retorts became used for distillations. Both alembics and retorts are forms of glassware with long necks pointing to the side at a downward angle to act as air-cooled condensers to condense the distillate and let it drip downward for collection. Later, copper alembics were invented. Riveted joints were often kept tight by using various mixtures, for instance a dough made of rye flour.[25] These alembics often featured a cooling system around the beak, using cold water, for instance, which made the condensation of alcohol more efficient. These were called pot stills. Today, the retorts and pot stills have been largely supplanted by more efficient distillation methods in most industrial processes. However, the pot still is still widely used for the elaboration of some fine alcohols, such as cognac, Scotch whisky, Irish whiskey, tequila, and some vodkas. Pot stills made of various materials (wood, clay, stainless steel) are also used by bootleggers in various countries. Small pot stills are also sold for use in the domestic production[26] of flower water or essential oils.
Early forms of distillation involved batch processes using one vaporization and one condensation. Purity was improved by further distillation of the condensate. Greater volumes were processed by simply repeating the distillation. Chemists reportedly carried out as many as 500 to 600 distillations in order to obtain a pure compound.[27]
In the early 19th century, the basics of modern techniques, including pre-heating and reflux, were developed.[27] In 1822, Anthony Perrier developed one of the first continuous stills, and then, in 1826, Robert Stein improved that design to make his patent still. In 1830, Aeneas Coffey got a patent for improving the design even further.[28] Coffey's continuous still may be regarded as the archetype of modern petrochemical units. The French engineer Armand Savalle developed his steam regulator around 1846.[29] In 1877, Ernest Solvay was granted a U.S. Patent for a tray column for ammonia distillation,[30] and the same and subsequent years saw developments in this theme for oils and spirits.
With the emergence of chemical engineering as a discipline at the end of the 19th century, scientific rather than empirical methods could be applied. The developing petroleum industry in the early 20th century provided the impetus for the development of accurate design methods, such as the McCabe–Thiele method by Ernest Thiele and the Fenske equation. The first industrial plant in the United States to use distillation as a means of ocean desalination opened in Freeport, Texas in 1961 with the hope of bringing water security to the region.[31] The availability of powerful computers has allowed direct computer simulations of distillation columns.
Applications of distillation
The application of distillation can roughly be divided into four groups: laboratory scale, industrial distillation, distillation of herbs for perfumery and medicinals (herbal distillate), and food processing. The latter two are distinctively different from the former two in that distillation is not used as a true purification method but more to transfer all volatiles from the source materials to the distillate in the processing of beverages and herbs.
The main difference between laboratory scale distillation and industrial distillation are that laboratory scale distillation is often performed on a batch basis, whereas industrial distillation often occurs continuously. In batch distillation, the composition of the source material, the vapors of the distilling compounds, and the distillate change during the distillation. In batch distillation, a still is charged (supplied) with a batch of feed mixture, which is then separated into its component fractions, which are collected sequentially from most volatile to less volatile, with the bottoms – remaining least or non-volatile fraction – removed at the end. The still can then be recharged and the process repeated.
In continuous distillation, the source materials, vapors, and distillate are kept at a constant composition by carefully replenishing the source material and removing fractions from both vapor and liquid in the system. This results in a more detailed control of the separation process.
Idealized distillation model
The boiling point of a liquid is the temperature at which the vapor pressure of the liquid equals the pressure around the liquid, enabling bubbles to form without being crushed. A special case is the normal boiling point, where the vapor pressure of the liquid equals the ambient atmospheric pressure.
It is a misconception that in a liquid mixture at a given pressure, each component boils at the boiling point corresponding to the given pressure, allowing the vapors of each component to collect separately and purely. However, this does not occur, even in an idealized system. Idealized models of distillation are essentially governed by Raoult's law and Dalton's law and assume that vapor–liquid equilibria are attained.
Raoult's law states that the vapor pressure of a solution is dependent on 1) the vapor pressure of each chemical component in the solution and 2) the fraction of solution each component makes up, a.k.a. the mole fraction. This law applies to ideal solutions, or solutions that have different components but whose molecular interactions are the same as or very similar to pure solutions.
Dalton's law states that the total pressure is the sum of the partial pressures of each individual component in the mixture. When a multi-component liquid is heated, the vapor pressure of each component will rise, thus causing the total vapor pressure to rise. When the total vapor pressure reaches the pressure surrounding the liquid, boiling occurs and liquid turns to gas throughout the bulk of the liquid. A mixture with a given composition has one boiling point at a given pressure when the components are mutually soluble. A mixture of constant composition does not have multiple boiling points.
An implication of one boiling point is that lighter components never cleanly "boil first". At boiling point, all volatile components boil, but for a component, its percentage in the vapor is the same as its percentage of the total vapor pressure. Lighter components have a higher partial pressure and, thus, are concentrated in the vapor, but heavier volatile components also have a (smaller) partial pressure and necessarily vaporize also, albeit at a lower concentration in the vapor. Indeed, batch distillation and fractionation succeed by varying the composition of the mixture. In batch distillation, the batch vaporizes, which changes its composition; in fractionation, liquid higher in the fractionation column contains more lights and boils at lower temperatures. Therefore, starting from a given mixture, it appears to have a boiling range instead of a boiling point, although this is because its composition changes: each intermediate mixture has its own, singular boiling point.
The idealized model is accurate in the case of chemically similar liquids, such as benzene and toluene. In other cases, severe deviations from Raoult's law and Dalton's law are observed, most famously in the mixture of ethanol and water. These compounds, when heated together, form an azeotrope, which is when the vapor phase and liquid phase contain the same composition. Although there are computational methods that can be used to estimate the behavior of a mixture of arbitrary components, the only way to obtain accurate vapor–liquid equilibrium data is by measurement.
It is not possible to completely purify a mixture of components by distillation, as this would require each component in the mixture to have a zero partial pressure. If ultra-pure products are the goal, then further chemical separation must be applied. When a binary mixture is vaporized and the other component, e.g., a salt, has zero partial pressure for practical purposes, the process is simpler.
Batch or differential distillation
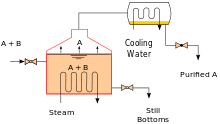
Heating an ideal mixture of two volatile substances, A and B, with A having the higher volatility, or lower boiling point, in a batch distillation setup (such as in an apparatus depicted in the opening figure) until the mixture is boiling results in a vapor above the liquid that contains a mixture of A and B. The ratio between A and B in the vapor will be different from the ratio in the liquid. The ratio in the liquid will be determined by how the original mixture was prepared, while the ratio in the vapor will be enriched in the more volatile compound, A (due to Raoult's Law, see above). The vapor goes through the condenser and is removed from the system. This, in turn, means that the ratio of compounds in the remaining liquid is now different from the initial ratio (i.e., more enriched in B than in the starting liquid).
The result is that the ratio in the liquid mixture is changing, becoming richer in component B. This causes the boiling point of the mixture to rise, which results in a rise in the temperature in the vapor, which results in a changing ratio of A : B in the gas phase (as distillation continues, there is an increasing proportion of B in the gas phase). This results in a slowly changing ratio of A : B in the distillate.
If the difference in vapour pressure between the two components A and B is large – generally expressed as the difference in boiling points – the mixture in the beginning of the distillation is highly enriched in component A, and when component A has distilled off, the boiling liquid is enriched in component B.
Continuous distillation
Continuous distillation is an ongoing distillation in which a liquid mixture is continuously (without interruption) fed into the process and separated fractions are removed continuously as output streams occur over time during the operation. Continuous distillation produces a minimum of two output fractions, including at least one volatile distillate fraction, which has boiled and been separately captured as a vapor and then condensed to a liquid. There is always a bottoms (or residue) fraction, which is the least volatile residue that has not been separately captured as a condensed vapor.
Continuous distillation differs from batch distillation in the respect that concentrations should not change over time. Continuous distillation can be run at a steady state for an arbitrary amount of time. For any source material of specific composition, the main variables that affect the purity of products in continuous distillation are the reflux ratio and the number of theoretical equilibrium stages, in practice determined by the number of trays or the height of packing. Reflux is a flow from the condenser back to the column, which generates a recycle that allows a better separation with a given number of trays. Equilibrium stages are ideal steps where compositions achieve vapor–liquid equilibrium, repeating the separation process and allowing better separation given a reflux ratio. A column with a high reflux ratio may have fewer stages, but it refluxes a large amount of liquid, giving a wide column with a large holdup. Conversely, a column with a low reflux ratio must have a large number of stages, thus requiring a taller column.
General improvements
Both batch and continuous distillations can be improved by making use of a fractionating column on top of the distillation flask. The column improves separation by providing a larger surface area for the vapor and condensate to come into contact. This helps it remain at equilibrium for as long as possible. The column can even consist of small subsystems ('trays' or 'dishes') which all contain an enriched, boiling liquid mixture, all with their own vapor–liquid equilibrium.
There are differences between laboratory-scale and industrial-scale fractionating columns, but the principles are the same. Examples of laboratory-scale fractionating columns (in increasing efficiency) include
- Air condenser
- Vigreux column (usually laboratory scale only)
- Packed column (packed with glass beads, metal pieces, or other chemically inert material)
- Spinning band distillation system.
Laboratory scale distillation
Laboratory scale distillations are almost exclusively run as batch distillations. The device used in distillation, sometimes referred to as a still, consists at a minimum of a reboiler or pot in which the source material is heated, a condenser in which the heated vapor is cooled back to the liquid state, and a receiver in which the concentrated or purified liquid, called the distillate, is collected. Several laboratory scale techniques for distillation exist (see also distillation types).
A completely sealed distillation apparatus could experience extreme and rapidly varying internal pressure, which could cause it to burst open at the joints. Therefore, some path is usually left open (for instance, at the receiving flask) to allow the internal pressure to equalize with atmospheric pressure. Alternatively, a vacuum pump may be used to keep the apparatus at a lower than atmospheric pressure. If the substances involved are air- or moisture-sensitive, the connection to the atmosphere can be made through one or more drying tubes packed with materials that scavenge the undesired air components, or through bubblers that provide a movable liquid barrier. Finally, the entry of undesired air components can be prevented by pumping a low but steady flow of suitable inert gas, like nitrogen, into the apparatus.
Simple distillation
In simple distillation, the vapor is immediately channeled into a condenser. Consequently, the distillate is not pure but rather its composition is identical to the composition of the vapors at the given temperature and pressure. That concentration follows Raoult's law.
As a result, simple distillation is effective only when the liquid boiling points differ greatly (rule of thumb is 25 °C)[32] or when separating liquids from non-volatile solids or oils. For these cases, the vapor pressures of the components are usually different enough that the distillate may be sufficiently pure for its intended purpose.
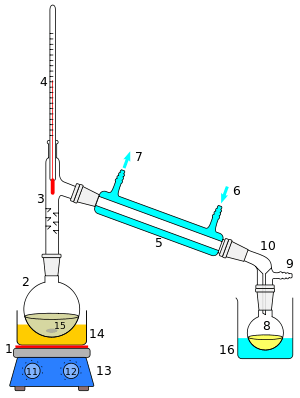
A cutaway schematic of a simple distillation operation is shown at left. The starting liquid 15 in the boiling flask 2 is heated by a combined hotplate and magnetic stirrer 13 via a silicone oil bath (orange, 14). The vapor flows through a short Vigreux column 3, then through a Liebig condenser 5, is cooled by water (blue) that circulates through ports 6 and 7. The condensed liquid drips into the receiving flask 8, sitting in a cooling bath (blue, 16). The adapter 10 has a connection 9 that may be fitted to a vacuum pump. The components are connected by ground glass joints (gray).
Fractional distillation

For many cases, the boiling points of the components in the mixture will be sufficiently close that Raoult's law must be taken into consideration. Therefore, fractional distillation must be used in order to separate the components by repeated vaporization-condensation cycles within a packed fractionating column. This separation, by successive distillations, is also referred to as rectification.[33]
As the solution to be purified is heated, its vapors rise to the fractionating column. As it rises, it cools, condensing on the condenser walls and the surfaces of the packing material. Here, the condensate continues to be heated by the rising hot vapors; it vaporizes once more. However, the composition of the fresh vapors are determined once again by Raoult's law. Each vaporization-condensation cycle (called a theoretical plate) will yield a purer solution of the more volatile component.[34] In reality, each cycle at a given temperature does not occur at exactly the same position in the fractionating column; theoretical plate is thus a concept rather than an accurate description.
More theoretical plates lead to better separations. A spinning band distillation system uses a spinning band of Teflon or metal to force the rising vapors into close contact with the descending condensate, increasing the number of theoretical plates.[35]
Steam distillation
Like vacuum distillation, steam distillation is a method for distilling compounds which are heat-sensitive.[36] The temperature of the steam is easier to control than the surface of a heating element, and allows a high rate of heat transfer without heating at a very high temperature. This process involves bubbling steam through a heated mixture of the raw material. By Raoult's law, some of the target compound will vaporize (in accordance with its partial pressure). The vapor mixture is cooled and condensed, usually yielding a layer of oil and a layer of water.
Steam distillation of various aromatic herbs and flowers can result in two products; an essential oil as well as a watery herbal distillate. The essential oils are often used in perfumery and aromatherapy while the watery distillates have many applications in aromatherapy, food processing and skin care.

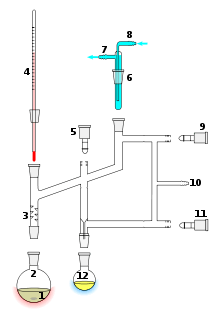
1: Stirrer bar/anti-bumping granules 2: Still pot 3: Fractionating column 4: Thermometer/Boiling point temperature 5: Teflon tap 1 6: Cold finger 7: Cooling water out 8: Cooling water in 9: Teflon tap 2 10: Vacuum/gas inlet 11: Teflon tap 3 12: Still receiver
Vacuum distillation
Some compounds have very high boiling points. To boil such compounds, it is often better to lower the pressure at which such compounds are boiled instead of increasing the temperature. Once the pressure is lowered to the vapor pressure of the compound (at the given temperature), boiling and the rest of the distillation process can commence. This technique is referred to as vacuum distillation and it is commonly found in the laboratory in the form of the rotary evaporator.
This technique is also very useful for compounds which boil beyond their decomposition temperature at atmospheric pressure and which would therefore be decomposed by any attempt to boil them under atmospheric pressure.
Molecular distillation is vacuum distillation below the pressure of 0.01 torr. 0.01 torr is one order of magnitude above high vacuum, where fluids are in the free molecular flow regime, i.e. the mean free path of molecules is comparable to the size of the equipment. The gaseous phase no longer exerts significant pressure on the substance to be evaporated, and consequently, rate of evaporation no longer depends on pressure. That is, because the continuum assumptions of fluid dynamics no longer apply, mass transport is governed by molecular dynamics rather than fluid dynamics. Thus, a short path between the hot surface and the cold surface is necessary, typically by suspending a hot plate covered with a film of feed next to a cold plate with a line of sight in between. Molecular distillation is used industrially for purification of oils.
Air-sensitive vacuum distillation
Some compounds have high boiling points as well as being air sensitive. A simple vacuum distillation system as exemplified above can be used, whereby the vacuum is replaced with an inert gas after the distillation is complete. However, this is a less satisfactory system if one desires to collect fractions under a reduced pressure. To do this a "cow" or "pig" adaptor can be added to the end of the condenser, or for better results or for very air sensitive compounds a Perkin triangle apparatus can be used.
The Perkin triangle, has means via a series of glass or Teflon taps to allows fractions to be isolated from the rest of the still, without the main body of the distillation being removed from either the vacuum or heat source, and thus can remain in a state of reflux. To do this, the sample is first isolated from the vacuum by means of the taps, the vacuum over the sample is then replaced with an inert gas (such as nitrogen or argon) and can then be stoppered and removed. A fresh collection vessel can then be added to the system, evacuated and linked back into the distillation system via the taps to collect a second fraction, and so on, until all fractions have been collected.
Short path distillation
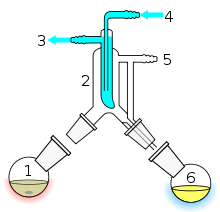
Short path distillation is a distillation technique that involves the distillate travelling a short distance, often only a few centimeters, and is normally done at reduced pressure.[37] A classic example would be a distillation involving the distillate travelling from one glass bulb to another, without the need for a condenser separating the two chambers. This technique is often used for compounds which are unstable at high temperatures or to purify small amounts of compound. The advantage is that the heating temperature can be considerably lower (at reduced pressure) than the boiling point of the liquid at standard pressure, and the distillate only has to travel a short distance before condensing. A short path ensures that little compound is lost on the sides of the apparatus. The Kugelrohr is a kind of a short path distillation apparatus which often contain multiple chambers to collect distillate fractions.
Zone distillation
Zone distillation is a distillation process in long container with partial melting of refined matter in moving liquid zone and condensation of vapor in the solid phase at condensate pulling in cold area. The process is worked in theory. When zone heater is moving from the top to the bottom of the container then solid condensate with irregular impurity distribution is forming. Then most pure part of the condensate may be extracted as product. The process may be iterated many times by moving (without turnover) the received condensate to the bottom part of the container on the place of refined matter. The irregular impurity distribution in the condensate (that is efficiency of purification) increases with the number of iterations. Zone distillation is the distillation analog of zone recrystallization. Impurity distribution in the condensate is described by known equations of zone recrystallization – with the replacement of the distribution co-efficient k of crystallization - for the separation factor α of distillation.[38][39][40]
Other types
- The process of reactive distillation involves using the reaction vessel as the still. In this process, the product is usually significantly lower-boiling than its reactants. As the product is formed from the reactants, it is vaporized and removed from the reaction mixture. This technique is an example of a continuous vs. a batch process; advantages include less downtime to charge the reaction vessel with starting material, and less workup. Distillation "over a reactant" could be classified as a reactive distillation. It is typically used to remove volatile impurity from the distallation feed. For example, a little lime may be added to remove carbon dioxide from water followed by a second distillation with a little sulfuric acid added to remove traces of ammonia.
- Catalytic distillation is the process by which the reactants are catalyzed while being distilled to continuously separate the products from the reactants. This method is used to assist equilibrium reactions reach completion.
- Pervaporation is a method for the separation of mixtures of liquids by partial vaporization through a non-porous membrane.
- Extractive distillation is defined as distillation in the presence of a miscible, high boiling, relatively non-volatile component, the solvent, that forms no azeotrope with the other components in the mixture.
- Flash evaporation (or partial evaporation) is the partial vaporization that occurs when a saturated liquid stream undergoes a reduction in pressure by passing through a throttling valve or other throttling device. This process is one of the simplest unit operations, being equivalent to a distillation with only one equilibrium stage.
- Codistillation is distillation which is performed on mixtures in which the two compounds are not miscible. In the laboratory, the Dean-Stark apparatus is used for this purpose to remove water from synthesis products. The Bleidner is another example with two refluxing solvents.
- Membrane distillation is a type of distillation in which vapors of a mixture to be separated are passed through a membrane, which selectively permeates one component of mixture. Vapor pressure difference is the driving force. It has potential applications in seawater desalination and in removal of organic and inorganic components.
The unit process of evaporation may also be called "distillation":
- In rotary evaporation a vacuum distillation apparatus is used to remove bulk solvents from a sample. Typically the vacuum is generated by a water aspirator or a membrane pump.
- In a kugelrohr a short path distillation apparatus is typically used (generally in combination with a (high) vacuum) to distill high boiling (> 300 °C) compounds. The apparatus consists of an oven in which the compound to be distilled is placed, a receiving portion which is outside of the oven, and a means of rotating the sample. The vacuum is normally generated by using a high vacuum pump.
Other uses:
- Dry distillation or destructive distillation, despite the name, is not truly distillation, but rather a chemical reaction known as pyrolysis in which solid substances are heated in an inert or reducing atmosphere and any volatile fractions, containing high-boiling liquids and products of pyrolysis, are collected. The destructive distillation of wood to give methanol is the root of its common name – wood alcohol.
- Freeze distillation is an analogous method of purification using freezing instead of evaporation. It is not truly distillation, but a recrystallization where the product is the mother liquor, and does not produce products equivalent to distillation. This process is used in the production of ice beer and ice wine to increase ethanol and sugar content, respectively. It is also used to produce applejack. Unlike distillation, freeze distillation concentrates poisonous congeners rather than removing them; As a result, many countries prohibit such applejack as a health measure. Also, distillation by evaporation can separate these since they have different boiling points.
Azeotropic distillation
Interactions between the components of the solution create properties unique to the solution, as most processes entail nonideal mixtures, where Raoult's law does not hold. Such interactions can result in a constant-boiling azeotrope which behaves as if it were a pure compound (i.e., boils at a single temperature instead of a range). At an azeotrope, the solution contains the given component in the same proportion as the vapor, so that evaporation does not change the purity, and distillation does not effect separation. For example, ethyl alcohol and water form an azeotrope of 95.6% at 78.1 °C.
If the azeotrope is not considered sufficiently pure for use, there exist some techniques to break the azeotrope to give a pure distillate. This set of techniques are known as azeotropic distillation. Some techniques achieve this by "jumping" over the azeotropic composition (by adding another component to create a new azeotrope, or by varying the pressure). Others work by chemically or physically removing or sequestering the impurity. For example, to purify ethanol beyond 95%, a drying agent (or desiccant, such as potassium carbonate) can be added to convert the soluble water into insoluble water of crystallization. Molecular sieves are often used for this purpose as well.
Immiscible liquids, such as water and toluene, easily form azeotropes. Commonly, these azeotropes are referred to as a low boiling azeotrope because the boiling point of the azeotrope is lower than the boiling point of either pure component. The temperature and composition of the azeotrope is easily predicted from the vapor pressure of the pure components, without use of Raoult's law. The azeotrope is easily broken in a distillation set-up by using a liquid–liquid separator (a decanter) to separate the two liquid layers that are condensed overhead. Only one of the two liquid layers is refluxed to the distillation set-up.
High boiling azeotropes, such as a 20 weight percent mixture of hydrochloric acid in water, also exist. As implied by the name, the boiling point of the azeotrope is greater than the boiling point of either pure component.
To break azeotropic distillations and cross distillation boundaries, such as in the DeRosier Problem, it is necessary to increase the composition of the light key in the distillate.
Breaking an azeotrope with unidirectional pressure manipulation
The boiling points of components in an azeotrope overlap to form a band. By exposing an azeotrope to a vacuum or positive pressure, it's possible to bias the boiling point of one component away from the other by exploiting the differing vapor pressure curves of each; the curves may overlap at the azeotropic point, but are unlikely to be remain identical further along the pressure axis either side of the azeotropic point. When the bias is great enough, the two boiling points no longer overlap and so the azeotropic band disappears.
This method can remove the need to add other chemicals to a distillation, but it has two potential drawbacks.
Under negative pressure, power for a vacuum source is needed and the reduced boiling points of the distillates requires that the condenser be run cooler to prevent distillate vapors being lost to the vacuum source. Increased cooling demands will often require additional energy and possibly new equipment or a change of coolant.
Alternatively, if positive pressures are required, standard glassware can not be used, energy must be used for pressurization and there is a higher chance of side reactions occurring in the distillation, such as decomposition, due to the higher temperatures required to effect boiling.
A unidirectional distillation will rely on a pressure change in one direction, either positive or negative.
Pressure-swing distillation
Pressure-swing distillation is essentially the same as the unidirectional distillation used to break azeotropic mixtures, but here both positive and negative pressures may be employed.
This improves the selectivity of the distillation and allows a chemist to optimize distillation by avoiding extremes of pressure and temperature that waste energy. This is particularly important in commercial applications.
One example of the application of pressure-swing distillation is during the industrial purification of ethyl acetate after its catalytic synthesis from ethanol.
Industrial distillation

Large scale industrial distillation applications include both batch and continuous fractional, vacuum, azeotropic, extractive, and steam distillation. The most widely used industrial applications of continuous, steady-state fractional distillation are in petroleum refineries, petrochemical and chemical plants and natural gas processing plants.
To control and optimize such industrial distillation, a standardized laboratory method, ASTM D86, is established. This test method extends to the atmospheric distillation of petroleum products using a laboratory batch distillation unit to quantitatively determine the boiling range characteristics of petroleum products.
Industrial distillation[33][41] is typically performed in large, vertical cylindrical columns known as distillation towers or distillation columns with diameters ranging from about 65 centimeters to 16 meters and heights ranging from about 6 meters to 90 meters or more. When the process feed has a diverse composition, as in distilling crude oil, liquid outlets at intervals up the column allow for the withdrawal of different fractions or products having different boiling points or boiling ranges. The "lightest" products (those with the lowest boiling point) exit from the top of the columns and the "heaviest" products (those with the highest boiling point) exit from the bottom of the column and are often called the bottoms.
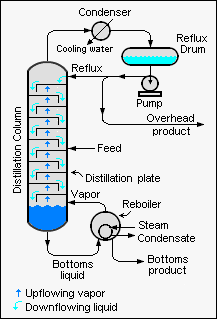
Industrial towers use reflux to achieve a more complete separation of products. Reflux refers to the portion of the condensed overhead liquid product from a distillation or fractionation tower that is returned to the upper part of the tower as shown in the schematic diagram of a typical, large-scale industrial distillation tower. Inside the tower, the downflowing reflux liquid provides cooling and condensation of the upflowing vapors thereby increasing the efficiency of the distillation tower. The more reflux that is provided for a given number of theoretical plates, the better the tower's separation of lower boiling materials from higher boiling materials. Alternatively, the more reflux that is provided for a given desired separation, the fewer the number of theoretical plates required. Chemical engineers must choose what combination of reflux rate and number of plates is both economically and physically feasible for the products purified in the distillation column.
Such industrial fractionating towers are also used in cryogenic air separation, producing liquid oxygen, liquid nitrogen, and high purity argon. Distillation of chlorosilanes also enables the production of high-purity silicon for use as a semiconductor.
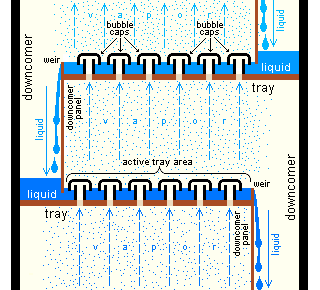
Design and operation of a distillation tower depends on the feed and desired products. Given a simple, binary component feed, analytical methods such as the McCabe–Thiele method[33][42] or the Fenske equation[33] can be used. For a multi-component feed, simulation models are used both for design and operation. Moreover, the efficiencies of the vapor–liquid contact devices (referred to as "plates" or "trays") used in distillation towers are typically lower than that of a theoretical 100% efficient equilibrium stage. Hence, a distillation tower needs more trays than the number of theoretical vapor–liquid equilibrium stages. A variety of models have been postulated to estimate tray efficiencies.
In modern industrial uses, a packing material is used in the column instead of trays when low pressure drops across the column are required. Other factors that favor packing are: vacuum systems, smaller diameter columns, corrosive systems, systems prone to foaming, systems requiring low liquid holdup, and batch distillation. Conversely, factors that favor plate columns are: presence of solids in feed, high liquid rates, large column diameters, complex columns, columns with wide feed composition variation, columns with a chemical reaction, absorption columns, columns limited by foundation weight tolerance, low liquid rate, large turn-down ratio and those processes subject to process surges.

This packing material can either be random dumped packing (1–3" wide) such as Raschig rings or structured sheet metal. Liquids tend to wet the surface of the packing and the vapors pass across this wetted surface, where mass transfer takes place. Unlike conventional tray distillation in which every tray represents a separate point of vapor–liquid equilibrium, the vapor–liquid equilibrium curve in a packed column is continuous. However, when modeling packed columns, it is useful to compute a number of "theoretical stages" to denote the separation efficiency of the packed column with respect to more traditional trays. Differently shaped packings have different surface areas and void space between packings. Both of these factors affect packing performance.
Another factor in addition to the packing shape and surface area that affects the performance of random or structured packing is the liquid and vapor distribution entering the packed bed. The number of theoretical stages required to make a given separation is calculated using a specific vapor to liquid ratio. If the liquid and vapor are not evenly distributed across the superficial tower area as it enters the packed bed, the liquid to vapor ratio will not be correct in the packed bed and the required separation will not be achieved. The packing will appear to not be working properly. The height equivalent to a theoretical plate (HETP) will be greater than expected. The problem is not the packing itself but the mal-distribution of the fluids entering the packed bed. Liquid mal-distribution is more frequently the problem than vapor. The design of the liquid distributors used to introduce the feed and reflux to a packed bed is critical to making the packing perform to it maximum efficiency. Methods of evaluating the effectiveness of a liquid distributor to evenly distribute the liquid entering a packed bed can be found in references.[44][45] Considerable work has been done on this topic by Fractionation Research, Inc. (commonly known as FRI).[46]
Multi-effect distillation
The goal of multi-effect distillation is to increase the energy efficiency of the process, for use in desalination, or in some cases one stage in the production of ultrapure water. The number of effects is inversely proportional to the kW·h/m3 of water recovered figure, and refers to the volume of water recovered per unit of energy compared with single-effect distillation. One effect is roughly 636 kW·h/m3.
- Multi-stage flash distillation can achieve more than 20 effects with thermal energy input, as mentioned in the article.
- Vapor compression evaporation – Commercial large-scale units can achieve around 72 effects with electrical energy input, according to manufacturers.
There are many other types of multi-effect distillation processes, including one referred to as simply multi-effect distillation (MED), in which multiple chambers, with intervening heat exchangers, are employed.
Distillation in food processing
Distilled beverages
Carbohydrate-containing plant materials are allowed to ferment, producing a dilute solution of ethanol in the process. Spirits such as whiskey and rum are prepared by distilling these dilute solutions of ethanol. Components other than ethanol, including water, esters, and other alcohols, are collected in the condensate, which account for the flavor of the beverage. Some of these beverages are then stored in barrels or other containers to acquire more flavor compounds and characteristic flavors.
Gallery
 | Chemistry in its beginnings used retorts as laboratory equipment exclusively for distillation processes. |
 | A simple set-up to distill dry and oxygen-free toluene. |
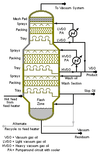 | Diagram of an industrial-scale vacuum distillation column as commonly used in oil refineries |
 | A rotary evaporator is able to distill solvents more quickly at lower temperatures through the use of a vacuum. |
 | Distillation using semi-microscale apparatus. The jointless design eliminates the need to fit pieces together. The pear-shaped flask allows the last drop of residue to be removed, compared with a similarly-sized round-bottom flask The small holdup volume prevents losses. A pig is used to channel the various distillates into three receiving flasks. If necessary the distillation can be carried out under vacuum using the vacuum adapter at the pig. |
See also
References
- Harwood & Moody 1989, pp. 141–143
- Schaschke, C., 2014. A Dictionary of Chemical Engineering. Oxford University Press.
- 2019. Distillation: The Historical Symbol of Chemical Engineering. The University of Toledo. URL https://www.utoledo.edu/engineering/chemical-engineering/distillation.html
- 2017. Products made from petroleum. Ranken Energy Corporation. URL https://www.ranken-energy.com/index.php/products-made-from-petroleum/
- Gildemeister, E.; Hoffman, Fr.; translated by Edward Kremers (1913). The Volatile Oils. 1. New York: Wiley. p. 203.
- Bryan H. Bunch; Alexander Hellemans (2004). The History of Science and Technology. Houghton Mifflin Harcourt. p. 88. ISBN 978-0-618-22123-3.
- Berthelot, Marcelin (1887–1888) Collection des anciens alchimistes grecs. 3 vol., Paris, p. 161
- Levey, Martin (1959). Chemistry and Chemical Technology in Ancient Mesopotamia. Elsevier. p. 36.
As already mentioned, the textual evidence for Sumero-Babylonian distillation is disclosed in a group of Akkadian tablets describing perfumery operations, dated ca. 1200 B.C.
- Forbes 1970, pp. 57, 89
- Taylor, F. (1945). "The evolution of the still". Annals of Science. 5 (3): 185. doi:10.1080/00033794500201451.
- Berthelot, M. P. E. M. (1893). "The Discovery of Alcohol and Distillation". The Popular Science Monthly. XLIII: 85–94. Archived from the original on 29 November 2017.
- John Marshall, Taxila, 2:420, 1951
- Frank Raymond Allchin, "India: the ancient home of distillation?" Man, New Series 14:1:55-63 (1979) full text
- Javed Husain, "The So-Called 'Distillery' at Shaikhan Dheri - A Case Study", Journal of the Pakistan Historical Society 41:3:289-314 (Jul 1, 1993)
- Habib, Irfan (2011), Economic History of Medieval India, 1200–1500. Pearson Education. p. 55. ISBN 9788131727911
- Haw, Stephen G. (2012). "Wine, women and poison". Marco Polo in China. Routledge. pp. 147–148. ISBN 978-1-134-27542-7.
The earliest possible period seems to be the Eastern Han dynasty ... the most likely period for the beginning of true distillation of spirits for drinking in China is during the Jin and Southern Song dynasties
- al-Hassan, Ahmad Y. (2001), Science and Technology in Islam: Technology and applied sciences. UNESCO. pp. 65–69. ISBN 9789231038310
- Hassan, Ahmad Y. "Alcohol and the Distillation of Wine in Arabic Sources". History of Science and Technology in Islam. Archived from the original on 29 December 2015. Retrieved 19 April 2014.
- The Economist: "Liquid fire – The Arabs discovered how to distil alcohol. They still do it best, say some" Archived 22 October 2012 at the Wayback Machine December 18, 2003
- Sarton, George (1975). Introduction to the history of science. R. E. Krieger Pub. Co. p. 145. ISBN 978-0-88275-172-6.
- Holmyard, Eric John (1990). Alchemy. Courier Dover Publications. p. 53. ISBN 978-0-486-26298-7.
- Braunschweig, Hieronymus (1500). Liber de arte destillandi, de Simplicibus [The Book of the Art of Distillation] (in German).
- French, John (1651). The Art of Distillation. London: Richard Cotes.
- "Distillation". Industrial & Engineering Chemistry. 28 (6): 677. 1936. doi:10.1021/ie50318a015.
- Sealing Technique, accessed 16 November 2006.
- Traditional Alembic Pot Still Archived 21 November 2006 at the Wayback Machine, accessed 16 November 2006.
- Othmer, D. F. (1982) "Distillation – Some Steps in its Development", in W. F. Furter (ed) A Century of Chemical Engineering. ISBN 0-306-40895-3
- GB 5974, Coffey, A., "Apparatus for Brewing and Distilling", published 5 August 1830, issued 5 February 1831; image Archived 4 February 2017 at the Wayback Machine
- Forbes 1970, p. 323
- US 198699, Solvay, Ernest, "Improvement in the Ammonia-Soda Manufacture", published 2 June 1876, issued 25 December 1877
- "Making the Deserts Bloom: Harnessing nature to deliver us from drought, Distillations Podcast and transcript, Episode 239". Science History Institute. 19 March 2019. Retrieved 27 August 2019.
- ST07 Separation of liquid–liquid mixtures (solutions), DIDAC by IUPAC
- Perry, Robert H.; Green, Don W. (1984). Perry's Chemical Engineers' Handbook (6th ed.). McGraw-Hill. ISBN 978-0-07-049479-4.
- Fractional Distillation. fandm.edu
- Spinning Band Distillation Archived 25 August 2006 at the Wayback Machine. B/R Instrument Corporation (accessed 8 September 2006)
- Harwood & Moody 1989, pp. 151–153
- Harwood & Moody 1989, p. 150
- Kravchenko, A. I. (2011). "Зонная дистилляция: новый метод рафинирования" [Zone distillation: a new method of refining]. Problems of Atomic Science and Technology (in Russian). 6 (19): 24–26.
- Kravchenko, A. I. (2014). "Zone distillation: justification". Problems of Atomic Science and Technology. 1 (20): 64–65.
- Kravchenko, A. I. (2014). "Разработка перспективных схем зонной дистилляции" [Design of advanced processes of zone distillation]. Perspectivnye Materialy (in Russian) (7): 68–72.
- Kister, Henry Z. (1992). Distillation Design (1st ed.). McGraw-Hill. ISBN 978-0-07-034909-4.
- Seader, J. D.; Henley, Ernest J. (1998). Separation Process Principles. New York: Wiley. ISBN 978-0-471-58626-5.
- Energy Institute website page Archived 12 October 2007 at the Wayback Machine. Resources.schoolscience.co.uk. Retrieved on 2014-04-20.
- Moore, F., Rukovena, F. (August 1987) Random Packing, Vapor and Liquid Distribution: Liquid and gas distribution in commercial packed towers, Chemical Plants & Processing, Edition Europe, pp. 11–15
- Spiegel, L (2006). "A new method to assess liquid distributor quality". Chemical Engineering and Processing. 45 (11): 1011. doi:10.1016/j.cep.2006.05.003.
- Kunesh, John G.; Lahm, Lawrence; Yanagi, Takashi (1987). "Commercial scale experiments that provide insight on packed tower distributors". Industrial & Engineering Chemistry Research. 26 (9): 1845. doi:10.1021/ie00069a021.
Cited sources
- Forbes, R. J. (1970). A Short History of the Art of Distillation from the Beginnings up to the Death of Cellier Blumenthal. BRILL. ISBN 978-90-04-00617-1.CS1 maint: ref=harv (link)
- Harwood, Laurence M.; Moody, Christopher J. (1989). Experimental organic chemistry: Principles and Practice (Illustrated ed.). Oxford: Blackwell Scientific Publications. ISBN 978-0-632-02017-1.CS1 maint: ref=harv (link)
Further reading
- Allchin, F. R. (1979). "India: The Ancient Home of Distillation?". Man. 14 (1): 55–63. doi:10.2307/2801640. JSTOR 2801640.
- Needham, Joseph (1980). Science and Civilisation in China. Cambridge University Press. ISBN 0-521-08573-X.
- Geankoplis, Christie John (2003). Transport Processes and Separation Process Principles (4th ed.). Prentice Hall. ISBN 978-0-13-101367-4.
External links
| Look up distillation in Wiktionary, the free dictionary. |
| Wikimedia Commons has media related to Distillation. |
- Alcohol distillation
- Case Study: Petroleum Distillation
- "Binary Vapor-Liquid Equilibrium Data" (searchable database). Chemical Engineering Research Information Center. Retrieved 5 May 2007.
| Principles | | |
|---|---|---|
| Industrial processes | ||
| Laboratory methods | ||
| Techniques | ||