Beta oxidation
In biochemistry and metabolism, beta-oxidation is the catabolic process by which fatty acid molecules are broken down[1] in the cytosol in prokaryotes and in the mitochondria in eukaryotes to generate acetyl-CoA, which enters the citric acid cycle, and NADH and FADH2, which are co-enzymes used in the electron transport chain. It is named as such because the beta carbon of the fatty acid undergoes oxidation to a carbonyl group. Beta-oxidation is primarily facilitated by the mitochondrial trifunctional protein, an enzyme complex associated with the inner mitochondrial membrane, although very long chain fatty acids are oxidized in peroxisomes.
The overall reaction for one cycle of beta oxidation is:
- Cn-acyl-CoA + FAD + NAD+
+ H
2O + CoA → Cn-2-acyl-CoA + FADH
2 + NADH + H+
+ acetyl-CoA
Activation and membrane transport
Free fatty acids cannot penetrate any biological membrane due to their negative charge. Free fatty acids must cross the cell membrane through specific transport proteins, such as the SLC27 family fatty acid transport protein.[2][3] Once in the cytosol, the following processes bring fatty acids into the mitochondrial matrix so that beta-oxidation can take place.
- Long-chain-fatty-acid—CoA ligase catalyzes the reaction between a fatty acid with ATP to give a fatty acyl adenylate, plus inorganic pyrophosphate, which then reacts with free coenzyme A to give a fatty acyl-CoA ester and AMP.
- If the fatty acyl-CoA has a long chain, then the carnitine shuttle must be utilized:
- Acyl-CoA is transferred to the hydroxyl group of carnitine by carnitine palmitoyltransferase I, located on the cytosolic faces of the outer and inner mitochondrial membranes.
- Acyl-carnitine is shuttled inside by a carnitine-acylcarnitine translocase, as a carnitine is shuttled outside.
- Acyl-carnitine is converted back to acyl-CoA by carnitine palmitoyltransferase II, located on the interior face of the inner mitochondrial membrane. The liberated carnitine is shuttled back to the cytosol, as an acyl-carnitine is shuttled into the matrix.
- If the fatty acyl-CoA contains a short chain, these short-chain fatty acids can simply diffuse through the inner mitochondrial membrane.[4]
| step-1 | step-2 | step-3 | step-4 |
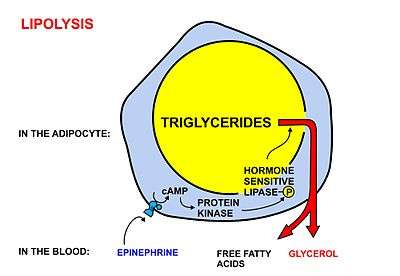 A diagrammatic illustration of the process of lipolysis (in a fat cell) induced by high epinephrine and low insulin levels in the blood. Epinephrine binds to a beta-adrenergic receptor in the cell wall of the adipocyte, which causes cAMP to be generated inside the cell. The cAMP activates a protein kinase, which phosphorylates and thus, in turn, activates a hormone-sensitive lipase in the fat cell. This lipase cleaves free fatty acids from their attachment to glycerol in the fat stored in the fat droplet of the adipocyte. The free fatty acids and glycerol are then released into the blood. | 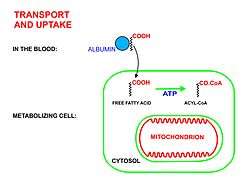 A diagrammatic illustration of the transport of free fatty acids in the blood attached to plasma albumin, its diffusion across the cell membrane using a protein transporter, and its activation, using ATP, to form acyl-CoA in the cytosol. The illustration is, for diagrammatic purposes, of a 12 carbon fatty acid. Most fatty acids in human plasma are 16 or 18 carbon atoms long. | 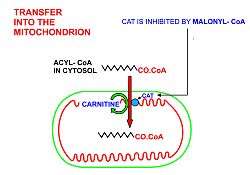 A diagrammatic illustration of the transfer of an acyl-CoA molecule across the inner membrane of the mitochondrion by carnitine-acyl-CoA transferase (CAT). The illustrated acyl chain is, for diagrammatic purposes, only 12 carbon atoms long. Most fatty acids in human plasma are 16 or 18 carbon atoms long. CAT is inhibited by high concentrations of malonyl-CoA (the first committed step in fatty acid synthesis) in the cytoplasm. This means that fatty acid synthesis and fatty acid catabolism cannot occur simultaneously in any given cell. | 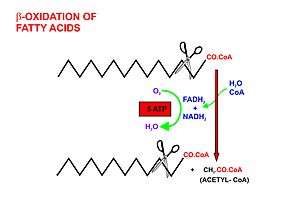 A diagrammatic illustration of the process of the beta-oxidation of an acyl-CoA molecule in the mitochodrial matrix. During this process an acyl-CoA molecule which is 2 carbons shorter than it was at the beginning of the process is formed. Acetyl-CoA, water and 5 ATP molecules are the other products of each beta-oxidative event, until the entire acyl-CoA molecule has been reduced to a set of acetyl-CoA molecules. |
General mechanism
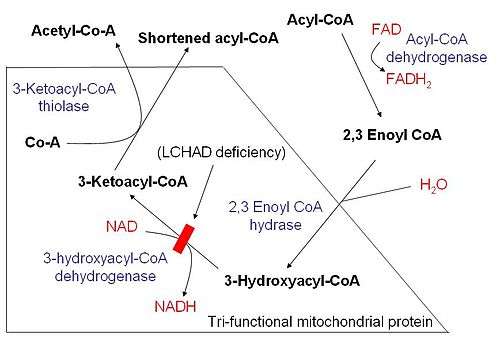
Once the fatty acid is inside the mitochondrial matrix, beta-oxidation occurs by cleaving two carbons every cycle to form acetyl-CoA. The process consists of 4 steps.
- A long-chain fatty acid is dehydrogenated to create a trans double bond between C2 and C3. This is catalyzed by acyl CoA dehydrogenase to produce trans-delta 2-enoyl CoA. It uses FAD as an electron acceptor and it is reduced to FADH2.
- Trans-delta2-enoyl CoA is hydrated at the double bond to produce L-3-hydroxyacyl CoA by enoyl-CoA hydratase.
- L-3-hydroxyacyl CoA is dehydrogenated again to create 3-ketoacyl CoA by 3-hydroxyacyl CoA dehydrogenase. This enzyme uses NAD as an electron acceptor.
- Thiolysis occurs between C2 and C3 (alpha and beta carbons) of 3-ketoacyl CoA. Thiolase enzyme catalyzes the reaction when a new molecule of coenzyme A breaks the bond by nucleophilic attack on C3. This releases the first two carbon units, as acetyl CoA, and a fatty acyl CoA minus two carbons. The process continues until all of the carbons in the fatty acid are turned into acetyl CoA.
Fatty acids are oxidized by most of the tissues in the body. However, some tissues such as the red blood cells of mammals (which do not contain mitochondria),[5] and cells of the central nervous system do not use fatty acids for their energy requirements,[6] but instead use carbohydrates (red blood cells and neurons) or ketone bodies (neurons only).[7][6]
Because many fatty acids are not fully saturated or do not have an even number of carbons, several different mechanisms have evolved, described below.
Even-numbered saturated fatty acids
Once inside the mitochondria, each cycle of β-oxidation, liberating a two carbon unit (acetyl-CoA), occurs in a sequence of four reactions:
| Description | Diagram | Enzyme | End product |
| Dehydrogenation by FAD: The first step is the oxidation of the fatty acid by Acyl-CoA-Dehydrogenase. The enzyme catalyzes the formation of a double bond between the C-2 and C-3. |  | acyl CoA dehydrogenase | trans-Δ2-enoyl-CoA |
| Hydration: The next step is the hydration of the bond between C-2 and C-3. The reaction is stereospecific, forming only the L isomer. | 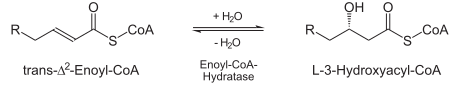 | enoyl CoA hydratase | L-β-hydroxyacyl CoA |
| Oxidation by NAD+: The third step is the oxidation of L-β-hydroxyacyl CoA by NAD+. This converts the hydroxyl group into a keto group. | 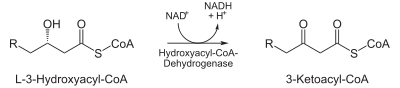 | 3-hydroxyacyl-CoA dehydrogenase | β-ketoacyl CoA |
| Thiolysis: The final step is the cleavage of β-ketoacyl CoA by the thiol group of another molecule of Coenzyme A. The thiol is inserted between C-2 and C-3. | 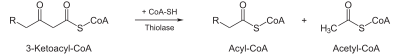 | β-ketothiolase | An acetyl-CoA molecule, and an acyl-CoA molecule that is two carbons shorter |
This process continues until the entire chain is cleaved into acetyl CoA units. The final cycle produces two separate acetyl CoAs, instead of one acyl CoA and one acetyl CoA. For every cycle, the Acyl CoA unit is shortened by two carbon atoms. Concomitantly, one molecule of FADH2, NADH and acetyl CoA are formed.
Odd-numbered saturated fatty acids
In general, fatty acids with an odd number of carbons are found in the lipids of plants and some marine organisms. Many ruminant animals form a large amount of 3-carbon propionate during the fermentation of carbohydrates in the rumen.[8] Long-chain fatty acids with an odd number of carbon atoms are found particularly in ruminant fat and milk.[9]
Chains with an odd-number of carbons are oxidized in the same manner as even-numbered chains, but the final products are propionyl-CoA and Acetyl CoA
Propionyl-CoA is first carboxylated using a bicarbonate ion into D-stereoisomer of methylmalonyl-CoA, in a reaction that involves a biotin co-factor, ATP, and the enzyme propionyl-CoA carboxylase. The bicarbonate ion's carbon is added to the middle carbon of propionyl-CoA, forming a D-methylmalonyl-CoA. However, the D conformation is enzymatically converted into the L conformation by methylmalonyl-CoA epimerase, then it undergoes intramolecular rearrangement, which is catalyzed by methylmalonyl-CoA mutase (requiring B12 as a coenzyme) to form succinyl-CoA. The succinyl-CoA formed can then enter the citric acid cycle.
However, whereas acetyl-CoA enters the citric acid cycle by condensing with an existing molecule of oxaloacetate, succinyl-CoA enters the cycle as a principal in its own right. Thus the succinate just adds to the population of circulating molecules in the cycle and undergoes no net metabolization while in it. When this infusion of citric acid cycle intermediates exceeds cataplerotic demand (such as for aspartate or glutamate synthesis), some of them can be extracted to the gluconeogenesis pathway, in the liver and kidneys, through phosphoenolpyruvate carboxykinase, and converted to free glucose.[10]
Unsaturated fatty acids
β-Oxidation of unsaturated fatty acids poses a problem since the location of a cis bond can prevent the formation of a trans-Δ2 bond. These situations are handled by an additional two enzymes, Enoyl CoA isomerase or 2,4 Dienoyl CoA reductase.
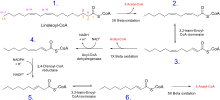
Whatever the conformation of the hydrocarbon chain, β-oxidation occurs normally until the acyl CoA (because of the presence of a double bond) is not an appropriate substrate for acyl CoA dehydrogenase, or enoyl CoA hydratase:
- If the acyl CoA contains a cis-Δ3 bond, then cis-Δ3-Enoyl CoA isomerase will convert the bond to a trans-Δ2 bond, which is a regular substrate.
- If the acyl CoA contains a cis-Δ4 double bond, then its dehydrogenation yields a 2,4-dienoyl intermediate, which is not a substrate for enoyl CoA hydratase. However, the enzyme 2,4 Dienoyl CoA reductase reduces the intermediate, using NADPH, into trans-Δ3-enoyl CoA. As in the above case, this compound is converted into a suitable intermediate by 3,2-Enoyl CoA isomerase.
To summarize:
- Odd-numbered double bonds are handled by the isomerase.
- Even-numbered double bonds by the reductase (which creates an odd-numbered double bond)
Peroxisomal beta-oxidation
Fatty acid oxidation also occurs in peroxisomes when the fatty acid chains are too long to be handled by the mitochondria. The same enzymes are used in peroxisomes as in the mitochondrial matrix, and acetyl-CoA is generated. It is believed that very long chain (greater than C-22) fatty acids, branched fatty acids,[11] some prostaglandins and leukotrienes[12] undergo initial oxidation in peroxisomes until octanoyl-CoA is formed, at which point it undergoes mitochondrial oxidation.[13]
One significant difference is that oxidation in peroxisomes is not coupled to ATP synthesis. Instead, the high-potential electrons are transferred to O2, which yields H2O2. It does generate heat however. The enzyme catalase, found primarily in peroxisomes and the cytosol of erythrocytes (and sometimes in mitochondria[14]), converts the hydrogen peroxide into water and oxygen.
Peroxisomal β-oxidation also requires enzymes specific to the peroxisome and to very long fatty acids. There are four key differences between the enzymes used for mitochondrial and peroxisomal β-oxidation:
- The NADH formed in the third oxidative step cannot be reoxidized in the peroxisome, so reducing equivalents are exported to the cytosol.
- β-oxidation in the peroxisome requires the use of a peroxisomal carnitine acyltransferase (instead of carnitine acyltransferase I and II used by the mitochondria) for transport of the activated acyl group into the mitochondria for further breakdown.
- The first oxidation step in the peroxisome is catalyzed by the enzyme acyl-CoA oxidase.
- The β-ketothiolase used in peroxisomal β-oxidation has an altered substrate specificity, different from the mitochondrial β-ketothiolase.
Peroxisomal oxidation is induced by a high-fat diet and administration of hypolipidemic drugs like clofibrate.
Energy yield
The ATP yield for every oxidation cycle is theoretically a maximum yield of 17, as NADH produces 3 ATP, FADH2 produces 2 ATP and a full rotation of the citric acid cycle produces 12 ATP. In practice it is closer to 14 ATP for a full oxidation cycle as the theoretical yield is not attained - it is generally closer to 2.5 ATP per NADH molecule produced, 1.5 ATP for each FADH2 molecule produced and this equates to 10 ATP per cycle of the TCA (according to the P/O ratio), broken down as follows:
| Source | ATP | Total |
| 1 FADH2 | x 1.5 ATP | = 1.5 ATP (Theoretically 2 ATP) |
| 1 NADH | x 2.5 ATP | = 2.5 ATP (Theoretically 3 ATP) |
| 1 acetyl CoA | x 10 ATP | = 10 ATP (Theoretically 12 ATP) |
| TOTAL | = 14 ATP |
For an even-numbered saturated fat (C2n), n - 1 oxidations are necessary, and the final process yields an additional acetyl CoA. In addition, two equivalents of ATP are lost during the activation of the fatty acid. Therefore, the total ATP yield can be stated as:
- (n - 1) * 14 + 10 - 2 = total ATP
or
- 14n-6 (alternatively)
For instance, the ATP yield of palmitate (C16, n = 8) is:
- (8 - 1) * 14 + 10 - 2 = 106 ATP
Represented in table form:
| Source | ATP | Total |
| 7 FADH2 | x 1.5 ATP | = 10.5 ATP |
| 7 NADH | x 2.5 ATP | = 17.5 ATP |
| 8 acetyl CoA | x 10 ATP | = 80 ATP |
| Activation | = -2 ATP | |
| NET | = 106 ATP |
For sources that use the larger ATP production numbers described above, the total would be 129 ATP ={(8-1)*17+12-2} equivalents per palmitate.
Beta-oxidation of unsaturated fatty acids changes the ATP yield due to the requirement of two possible additional enzymes.
Similarities between beta-oxidation and citric acid cycle
The reactions of beta oxidation and part of citric acid cycle present structural similarities in three of four reactions of the beta oxidation: the oxidation by FAD, the hydration, and the oxidation by NAD+. Each enzyme of these metabolic pathways presents structural similarity.
Clinical significance
There are at least 25 enzymes and specific transport proteins in the β-oxidation pathway.[15] Of these, 18 have been associated with human disease as inborn errors of metabolism.
See also
References
- Houten SM, Wanders RJ (October 2010). "A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation". Journal of Inherited Metabolic Disease. 33 (5): 469–77. doi:10.1007/s10545-010-9061-2. PMC 2950079. PMID 20195903.
- Stahl A (February 2004). "A current review of fatty acid transport proteins (SLC27)". Pflügers Archiv. 447 (5): 722–7. doi:10.1007/s00424-003-1106-z. PMID 12856180.
- Anderson CM, Stahl A (April 2013). "SLC27 fatty acid transport proteins". Molecular Aspects of Medicine. 34 (2–3): 516–28. doi:10.1016/j.mam.2012.07.010. PMC 3602789. PMID 23506886.
- Charney AN, Micic L, Egnor RW (March 1998). "Nonionic diffusion of short-chain fatty acids across rat colon". The American Journal of Physiology. 274 (3 Pt 1): G518–24. doi:10.1152/ajpgi.1998.274.3.G518. PMID 9530153.
- Stier A, Bize P, Schull Q, Zoll J, Singh F, Geny B, Gros F, Royer C, Massemin S, Criscuolo F (June 2013). "Avian erythrocytes have functional mitochondria, opening novel perspectives for birds as animal models in the study of ageing". Frontiers in Zoology. 10 (1): 33. doi:10.1186/1742-9994-10-33. PMC 3686644. PMID 23758841.
- Schönfeld P, Reiser G (October 2013). "Why does brain metabolism not favor burning of fatty acids to provide energy? Reflections on disadvantages of the use of free fatty acids as fuel for brain". Journal of Cerebral Blood Flow and Metabolism. 33 (10): 1493–9. doi:10.1038/jcbfm.2013.128. PMC 3790936. PMID 23921897.
- Yoshida T, Shevkoplyas SS (October 2010). "Anaerobic storage of red blood cells". Blood Transfusion = Trasfusione del Sangue. 8 (4): 220–36. doi:10.2450/2010.0022-10. PMC 2957487. PMID 20967163.
- Nelson DL, Cox MM (2005). Lehninger Principles of Biochemistry (4th ed.). New York: W. H. Freeman and Company. pp. 648–649. ISBN 978-0-7167-4339-2.
- Rodwell VW. Harper's Illustrated Biochemistry (31st ed.). McGraw-Hill Publishing Company.
- King M. "Gluconeogenesis: Synthesis of New Glucose". Subsection: "Propionate". themedicalbiochemistrypage.org, LLC. Retrieved 20 March 2013.
- Singh I (February 1997). "Biochemistry of peroxisomes in health and disease". Molecular and Cellular Biochemistry. 167 (1–2): 1–29. doi:10.1023/A:1006883229684. PMID 9059978.
- Gibson GG, Lake BG (2013-04-08). Peroxisomes: Biology and Importance in Toxicology and Medicine. CRC Press. pp. 69–. ISBN 978-0-203-48151-6.
- Lazarow PB (March 1978). "Rat liver peroxisomes catalyze the beta oxidation of fatty acids". The Journal of Biological Chemistry. 253 (5): 1522–8. PMID 627552.
- Bai J, Cederbaum AI (2001). "Mitochondrial catalase and oxidative injury". BIOLOGICAL SIGNALS AND RECEPTORS. 10 (3–4): 3189–199. PMID 11351128.
- Tein I (2013). Disorders of fatty acid oxidation. Handbook of Clinical Neurology. 113. pp. 1675–88. doi:10.1016/B978-0-444-59565-2.00035-6. ISBN 9780444595652. PMID 23622388.
Further reading
- Berg JM, Tymoczko JL, Stryer L (2002). "Certain Fatty Acids Require Additional Steps for Degradation". Biochemistry (5th ed.). New York: W H Freeman. ISBN 978-0-7167-4684-3.
External links
- Silva P. "The chemical logic behind fatty acid metabolism". Universidade Fernando Pessoa. Archived from the original on 16 March 2010.
- "Fatty acid oxidation animation". Cengage Learning.
Metabolism map | ||
|---|---|---|
Single lines: pathways common to most lifeforms. Double lines: pathways not in humans (occurs in e.g. plants, fungi, prokaryotes). | ||
.svg.png)