Systemic primary carnitine deficiency
Systemic primary carnitine deficiency (SPCD)[1] is an inborn error of fatty acid transport caused by a defect in the transporter responsible for moving carnitine across the plasma membrane. Carnitine is an important amino acid for fatty acid metabolism.[4] When carnitine cannot be transported into tissues, fatty acid oxidation is impaired, leading to a variety of symptoms such as chronic muscle weakness, cardiomyopathy, hypoglycemia and liver dysfunction. The specific transporter involved with SPCD is OCTN2, coded for by the SLC22A5 gene located on chromosome 5. SPCD is inherited in an autosomal recessive manner, with mutated alleles coming from both parents.
| Systemic primary carnitine deficiency | |
|---|---|
| Other names | Carnitine deficiency, systemic primary (CDSP),[1][2] Carnitine uptake defect (CUD),[1] Carnitine transporter deficiency (CTD)[3] or Systemic carnitine deficiency (SCD)[2] |
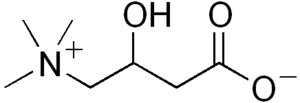 | |
| Carnitine | |
| Specialty | Endocrinology |
Acute episodes due to SPCD are often preceded by metabolic stress such as extended fasting, infections or vomiting. Cardiomyopathy can develop in the absence of an acute episode, and can result in death. SPCD leads to increased carnitine excretion in the urine and low levels in plasma. In most locations with expanded newborn screening, SPCD can be identified and treated shortly after birth. Treatment with high doses of carnitine supplementation is effective, but needs to be rigorously maintained for life.
SPCD is more common in the Faroe Islands than in other countries; at least one out of every 1000 inhabitants of the Faroes has the illness,[5] while the numbers for other countries are one in every 100,000. Around 100 persons in the islands have been diagnosed, around one third of the whole population of 48,000 people have been screened for SPCD. Several young Faroese people and children have died a sudden death with cardiac arrest because of SPCD. Scientists believe that around 10% of the Faroese population are carriers of variants which cause SPCD.[6] These people are not ill, but may have a lower amount of carnitine in their blood than non-carriers.
Signs and symptoms
The presentation of patient with SPCD can be incredibly varied, from asymptomatic to lethal cardiac manifestations.[7] Early cases were reported with liver dysfunction, muscular findings (weakness and underdevelopment), hypoketotic hypoglycemia, cardiomegaly, cardiomyopathy and marked carnitine deficiency in plasma and tissues, combined with increased excretion in urine.[7] Patients who present clinically with SPCD fall into two categories, a metabolic presentation with hypoglycemia and a cardiac presentation characterized by cardiomyopathy. Muscle weakness can be found with either presentation.[8]
In countries with expanded newborn screening, SPCD can be identified shortly after birth. Affected infants show low levels of free carnitine and all other acylcarnitine species by tandem mass spectrometry.[8] Not all infants with low free carnitine are affected with SPCD. Some may have carnitine deficiency secondary to another metabolic condition or due to maternal carnitine deficiency. Proper follow-up of newborn screening results for low free carnitine includes studies of the mother to determine whether her carnitine deficiency is due to SPCD or secondary to a metabolic disease or diet.[9] Maternal cases of SPCD have been identified at a higher than expected rate, often in women who are asymptomatic.[8][10] Some mothers have also been identified through newborn screening with cardiomyopathy that had not been previously diagnosed.[11] The identification and treatment of these asymptomatic individuals is still developing, as it is not clear whether they require the same levels of intervention as patients identified with SPCD early in life based on clinical presentation.[8]
Genetics
SPCD is an autosomal recessive condition, meaning a mutated allele must be inherited from each parent for an individual to be affected.[7] The gene responsible for the OCTN2 carnitine transporter is SLC22A5, located at 5q31.1-32. SLC22A5 is regulated by peroxisome proliferator-activated receptor alpha. The transporter, OCTN2, is located in the apical membrane of the renal tubular cells, where it plays a role in tubular reabsorption.[8]
The defective OCTN2 is unable to recapture carnitine prior to its excretion in urine, leading to the characteristic biochemical findings of massively increased urine carnitine levels and significantly decreased plasma carnitine levels.[7] Decreased levels of plasma carnitine inhibit fatty acid oxidation during times of excessive energy demand. Carnitine is needed to transport long chain fatty acids into the mitochondria, where they can be broken down to produce acetyl-CoA. Individuals with SPCD cannot produce ketone bodies as energy due to the interruption of fatty acid oxidation.[8] Although SPCD is an autosomal recessive condition, heterozygotes have been shown to be at an increased risk for developing benign cardiomyopathy compared to wild type individuals.[7]
Diagnosis
The first suspicion of SPCD in a patient with a non-specific presentation is an extremely low plasma carnitine level. When combined with an increased concentration of carnitine in urine, the suspicion of SPCD can often be confirmed by either molecular testing or functional studies assessing the uptake of carnitine in cultured fibroblasts.[8]
Treatment
Identification of patients presymptomatically via newborn screening has allowed early intervention and treatment. Treatment for SPCD involves high dose carnitine supplementation, which must be continued for life.[8] Individuals who are identified and treated at birth have very good outcomes, including the prevention of cardiomyopathy.[7] Mothers who are identified after a positive newborn screen but are otherwise asymptomatic are typically offered carnitine supplementation as well. The long-term outcomes for asymptomatic adults with SPCD is not known, but the discovery of mothers with undiagnosed cardiomyopathy and SPCD has raised the possibility that identification and treatment may prevent adult-onset manifestations.[8][11]
Incidence
The addition of SPCD to newborn screening panels has offered insight into the incidence of the disorder around the world. In Taiwan, the incidence of SPCD in newborns was estimated to be approximately 1:67,000, while maternal cases were identified at a higher frequency of approximately 1:33,000.[11] The increased incidence of SPCD in mothers compared to newborns is not completely understood.[8] Estimates of SPCD in Japan have shown a similar incidence of 1:40,000.[7] Worldwide, SPCD has the highest incidence in the relatively genetically isolated Faroe Islands, where an extensive screening program was instituted after the sudden death of two teenagers. The incidence in the Faroe Islands is approximately 1:200.[12][13]
History
Carnitine deficiency has been extensively studied, although most commonly as a secondary finding to other metabolic conditions.[7] The first case of SPCD was reported in the 1980s, in a child with fasting hypoketotic hypoglycemia that resolved after treatment with carnitine supplementation. Later cases were reported with cardiomyopathy and muscle weakness. Newborn screening expanded the potential phenotypes associated with SPCD, to include otherwise asymptomatic adults.[8]
References
- Systemic primary carnitine deficiency Orphanet
- Online Mendelian Inheritance in Man (OMIM): 212140
- Carnitine transporter deficiency - newbornscreening.info
- Activation and Transportation of Fatty Acids for Metabolism via Carnitine Shuttle
- Hmr.fo - Faroe Islands Ministry of Health - Information om CTD (Carnitin Transporter Defekt) Archived 2014-02-22 at the Wayback Machine
- Dr.dk - Livsfarlig sygdom angriber færinge - By Tine Maria Borresø, 17 May 2010
- "#212140; Carnitine Deficiency, Systemic Primary; SPCD". Johns Hopkins University. Retrieved 2012-06-03.
- Stanley, Charles A.; Bennett, Michael J.; Longo, Nicolo (2004). "Plasma Membrane Carnitine Transport Defect". In Scriver, C.W.; Beaudet, A.L.; Sly, W.S.; et al. (eds.). Metabolic and Molecular Bases of Inherited Disease (8th ed.). New York: McGraw Hill.
- "C0 Free Carnitine Low" (PDF). American College of Medical Genetics. Retrieved 2012-06-03.
- Morris, Andrew A.M.; Spiekerkoetter, Ute (2012). "Disorders of Mitochondrial Fatty Acid Oxidation and Related Metabolic Pathways". In Saudubray, Jean-Marie; van den Berghe, Georges; Walter, John H. (eds.). Inborn Metabolic Diseases: Diagnosis and Treatment (5th ed.). New York: Springer. pp. 201–216. ISBN 978-3-642-15719-6.
- Lee, N. C.; Tang, N. L. S.; Chien, Y. H.; Chen, C. A.; Lin, S. J.; Chiu, P. C.; Huang, A. C.; Hwu, W. L. (2010). "Diagnoses of newborns and mothers with carnitine uptake defects through newborn screening". Molecular Genetics and Metabolism. 100 (1): 46–50. doi:10.1016/j.ymgme.2009.12.015. PMID 20074989.
- "Rare genetic disease common in Faroe Islands: expert". Yahoo!. 2010-05-27. Retrieved 2012-06-03.
- Lund, A. M.; Joensen, F.; Hougaard, D. M.; Jensen, L. K.; Christensen, E.; Christensen, M.; Nørgaard-Petersen, B.; Schwartz, M.; Skovby, F. (2007). "Carnitine transporter and holocarboxylase synthetase deficiencies in the Faroe Islands". Journal of Inherited Metabolic Disease. 30 (3): 341–349. doi:10.1007/s10545-007-0527-9. PMID 17417720.
External links
| Classification |
|
|---|---|
| External resources |