Transforming growth factor beta
Transforming growth factor beta (TGF-β) is a multifunctional cytokine belonging to the transforming growth factor superfamily that includes three[1] different mammalian isoforms (TGF-β 1 to 3, HGNC symbols TGFB1, TGFB2, TGFB3) and many other signaling proteins. TGFB proteins are produced by all white blood cell lineages.
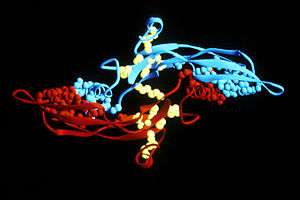
Activated TGF-β complexes with other factors to form a serine/threonine kinase complex that binds to TGF-β receptors. TGF-β receptors are composed of both type 1 and type 2 receptor subunits. After the binding of TGF-β, the type 2 receptor kinase phosphorylates and activates the type 1 receptor kinase that activates a signaling cascade.[2] This leads to the activation of different downstream substrates and regulatory proteins, inducing transcription of different target genes that function in differentiation, chemotaxis, proliferation, and activation of many immune cells.[2][3]
TGF-β is secreted by many cell types, including macrophages, in a latent form in which it is complexed with two other polypeptides, latent TGF-beta binding protein (LTBP) and latency-associated peptide (LAP). Serum proteinases such as plasmin catalyze the release of active TGF-β from the complex. This often occurs on the surface of macrophages where the latent TGF-β complex is bound to CD36 via its ligand, thrombospondin-1 (TSP-1). Inflammatory stimuli that activate macrophages enhance the release of active TGF-β by promoting the activation of plasmin. Macrophages can also endocytose IgG-bound latent TGF-β complexes that are secreted by plasma cells and then release active TGF-β into the extracellular fluid.[4] Among its key functions is regulation of inflammatory processes, particularly in the gut.[5] TGF-β also plays a crucial role in stem cell differentiation as well as T-cell regulation and differentiation.[6][7]
Because of its role in immune and stem cell regulation and differentiation, it is a highly researched cytokine in the fields of cancer, auto-immune diseases, and infectious disease.
The TGF-β superfamily includes endogenous growth inhibiting proteins; an increase in expression of TGF-β often correlates with the malignancy of many cancers and a defect in the cellular growth inhibition response to TGF-β. Its immunosuppressive functions then come to dominate, contributing to oncogenesis.[8] The dysregulation of its immunosuppressive functions is also implicated in the pathogenesis of autoimmune diseases, although their effect is mediated by the environment of other cytokines present.[5]
Structure
The primary 3 mammalian types are:
- TGF beta 1 – TGFB1[9][10]
- TGF beta 2 – TGFB2[11][12]
- TGF beta 3 – TGFB3[13][14]
A fourth member, TGF beta 4, has been identified in birds[15] – TGRB4 (synonyms: endometrial bleeding associated factor beta-4 (EBAF), Lefty preproprotein, LEFTA; Left-Right Determination Factor 2; LEFTYA; Left-Right Determination Factor A; Transforming Growth Factor Beta-4; Protein Lefty-2; Protein Lefty-A[16][17][18][19]).[20]
A fourth member of the subfamily, TGFB4, has been identified in birds and a fifth, TGFB5, only in frogs.[15]
The peptide structures of the TGF-β isoforms are highly similar (homologies on the order of 70–80%). They are all encoded as large protein precursors; TGF-β1 contains 390 amino acids and TGF-β2 and TGF-β3 each contain 412 amino acids. They each have an N-terminal signal peptide of 20–30 amino acids that they require for secretion from a cell, a pro-region called latency associated peptide (LAP - Alias: Pro-TGF beta 1, LAP/TGF beta 1), and a 112-114 amino acid C-terminal region that becomes the mature TGF-β molecule following its release from the pro-region by proteolytic cleavage.[21] The mature TGF-β protein dimerizes to produce a 25 KDa active protein with many conserved structural motifs.[22] TGF-β has nine cysteine residues that are conserved among its family. Eight form disulfide bonds within the protein to create a cysteine knot structure characteristic of the TGF-β superfamily. The ninth cysteine forms a disulfide bond with the ninth cysteine of another TGF-β protein to produce a dimer.[23] Many other conserved residues in TGF-β are thought to form secondary structure through hydrophobic interactions. The region between the fifth and sixth conserved cysteines houses the most divergent area of TGF-β proteins that is exposed at the surface of the protein and is implicated in receptor binding and specificity of TGF-β.
Latent TGF-β complex
All three TGF-βs are synthesized as precursor molecules containing a propeptide region in addition to the TGF-β homodimer.[24] After it is synthesized, the TGF-β homodimer interacts with a Latency Associated Peptide (LAP), a protein derived from the N-terminal region of the TGF-β gene product, forming a complex called Small Latent Complex (SLC). This complex remains in the cell until it is bound by another protein called Latent TGF-β-Binding Protein (LTBP), forming a larger complex called Large Latent Complex (LLC). It is this LLC that gets secreted to the extracellular matrix (ECM).[25]
In most cases, before the LLC is secreted, the TGF-β precursor is cleaved from the propeptide but remains attached to it by noncovalent bonds.[26] After its secretion, it remains in the extracellular matrix as an inactivated complex containing both the LTBP and the LAP which need to be further processed in order to release active TGF-β.[27] The attachment of TGF-β to the LTBP is by disulfide bond which allows it to remain inactive by preventing it from binding to its receptors . Because different cellular mechanisms require distinct levels of TGF-β signaling, the inactive complex of this cytokine gives opportunity for a proper mediation of TGF-β signaling.[27]
There are four different LTBP isoforms known, LTBP-1, LTBP-2, LTBP-3 and LTBP-4.[28] Mutation or alteration of LAP or LTBP can result in improper TGF-β signaling. Mice lacking LTBP-3 or LTBP-4 demonstrate phenotypes consistent to phenotypes seen in mice with altered TGF-β signaling.[29] Furthermore, specific LTBP isoforms have a propensity to associate with specific LAP•TGF-β isoforms. For example, LTBP-4 is reported to bind only to TGF-β1,[30] thus, mutation in LTBP-4 can lead to TGF-β associated complications which are specific to tissues that predominantly involves TGF-β1. Moreover, the structural differences within the LAP's provide different latent TGF-β complexes which are selective but to specific stimuli generated by specific activators.
Activation
Although TGF-β is important in regulating crucial cellular activities, only a few TGF-β activating pathways are currently known, and the full mechanism behind the suggested activation pathways is not yet well understood. Some of the known activating pathways are cell or tissue specific, while some are seen in multiple cell types and tissues.[27][31] Proteases, integrins, pH, and reactive oxygen species are just few of the currently known factors that can activate TGF-β, as discussed below.[32][33][34] It is well known that perturbations of these activating factors can lead to unregulated TGF-β signaling levels that may cause several complications including inflammation, autoimmune disorders, fibrosis, cancer and cataracts.[35][36] In most cases, an activated TGF-β ligand will initiate the TGF-β signaling cascade as long as TGF-β receptors I and II are available for binding. This is due to a high affinity between TGF-β and its receptors, suggesting why the TGF-β signaling recruits a latency system to mediate its signaling.[27]
Integrin-independent activation
Activation by protease and metalloprotease
Plasmin and a number of matrix metalloproteinases (MMP) play a key role in promoting tumor invasion and tissue remodeling by inducing proteolysis of several ECM components.[32] The TGF-β activation process involves the release of the LLC from the matrix, followed by further proteolysis of the LAP to release TGF-β to its receptors. MMP-9 and MMP-2 are known to cleave latent TGF-β.[35] The LAP complex contains a protease-sensitive hinge region which can be the potential target for this liberation of TGF-β.[36] Despite the fact that MMPs have been proven to play a key role in activating TGF-β, mice with mutations in MMP-9 and MMP-2 genes can still activate TGF-β and do not show any TGF-β deficiency phenotypes, this may reflect redundancy among the activating enzymes[27] suggesting that other unknown proteases might be involved.
Activation by pH
Acidic conditions can denature the LAP. Treatment of the medium with extremes of pH (1.5 or 12) resulted in significant activation of TGF-β as shown by radio-receptor assays, while mild acid treatment (pH 4.5) yielded only 20-30% of the activation achieved by pH 1.5.[37]
Activation by reactive oxygen species (ROS)
The structure of LAP is important in maintaining its function. Structure modification of LAP can lead to disturb the interaction between LAP and TGF-β and thus activating it. Factors that may cause such modification may include hydroxyl radicals from reactive oxygen species (ROS). TGF-β was rapidly activated after in vivo radiation exposure ROS.[33]
Activation by thrombospondin-1
Thrombospondin-1 (TSP-1) is a matricellular glycoprotein found in plasma of healthy patients with levels in the range of 50–250 ng/ml.[38] TSP-1 levels are known to increase in response to injury and during development.[39] TSP-1 activates latent TGF-beta[40] by forming direct interactions with the latent TGF-β complex and induces a conformational rearrangement preventing it from binding to the matured TGF-β.[41]
Activation by Alpha(V) containing integrins
The general theme of integrins participating in latent TGF-β1 activation arose from studies that examined mutations/knockouts of β6 integrin,[42] αV integrin,[43] β8 integrin and in LAP. These mutations produced phenotypes that were similar to phenotypes seen in TGF-β1 knockout mice.[44] Currently there are two proposed models of how αV containing integrins can activate latent TGF-β1; the first proposed model is by inducing conformational change to the latent TGF-β1 complex and hence releasing the active TGF-β1 and the second model is by a protease-dependent mechanism.[34]
Conformation change mechanism pathway (without proteolysis)
αVβ6 integrin was the first integrin to be identified as TGF-β1 activator.[27] LAPs contain an RGD motif which is recognized by vast majority of αV containing integrins,[45] and αVβ6 integrin can activate TGF-β1 by binding to the RGD motif present in LAP-β1 and LAP-β 3.[46] Upon binding, it induces adhesion-mediated cell forces that are translated into biochemical signals which can lead to liberation/activation of TGFb from its latent complex.[47] This pathway has been demonstrated for activation of TGF-β in epithelial cells and does not associate MMPs.[48]
Integrin protease-dependent activation mechanism
Because MMP-2 and MMP-9 can activate TGF-β through proteolytic degradation of the latent TGF beta complex,[35] αV containing integrins activate TGF-β1 by creating a close connection between the latent TGF-β complex and MMPs. Integrins αVβ6 and αVβ3 are suggested to simultaneously bind the latent TGF-β1 complex and proteinases, simultaneous inducing conformational changes of the LAP and sequestering proteases to close proximity. Regardless of involving MMPs, this mechanism still necessitate the association of integrins and that makes it a non proteolytic pathway.[34][49]
Signaling pathways
Canonical signaling: The SMAD pathway
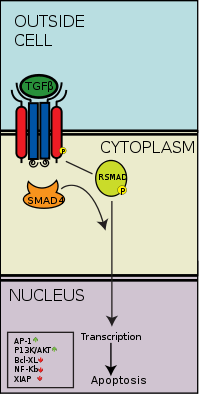
Smads are a class of intracellular signalling proteins and transcription factors for the TGF-β family of signalling molecules. This pathway conceptually resembles the Jak-STAT signal transduction pathway characterized in the activation of cytokine receptors implicated, for example, in the B cell isotype switching pathway. As previously stated, the binding of the TGF-β ligand to the TGF-β receptor, the type 2 receptor kinase phosphorylates and activates the type 1 receptor kinase that activates a signaling cascade. In the case of Smad, receptor-activated Smads are phosphorylated by the type 1 TGF-β receptor kinase, and these go on to complex with other Smads, which is able to translocate into the cell nucleus to induce transcription of different effectors.[50]
More specifically, activated TGF-β complexes bind to the type 2 domain of the TGF-β receptor which then recruits and phosphorylates a type 1 receptor. The type 1 receptor then recruits and phosphorylates a receptor regulated SMAD (R-SMAD). The R-SMAD then binds to the common SMAD (coSMAD) SMAD4 and forms a heterodimeric complex. This complex then enters the cell nucleus where it acts as a transcription factor for various genes, including those to activate the mitogen-activated protein kinase 8 pathway, which triggers apoptosis. The SMAD pathway is regulated by feedback inhibition. SMAD6 and SMAD7 may block type I receptors.[51] There is also substantial evidence that TGF-β-dependent signaling via the SMAD-3 pathway is responsible for many of the inhibitory functions of TGF-β discussed in later sections and thus it is implicated in oncogenesis.[52]
Apoptosis via the DAXX pathway
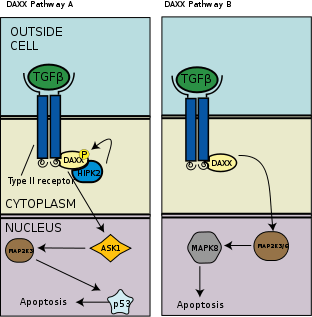
TGF-β induces apoptosis, or programmed cell death, in human lymphocytes and hepatocytes. The importance of this function is clear in TGF-β deficient mice which experience hyperproliferation and unregulated autoimmunity.[53] In a separate apoptotic pathway from the association of death-associated protein 6 (DAXX) with the death receptor Fas, there is evidence of association and binding between DAXX and type 2 TGF-β receptor kinase, wherein DAXX binds to the C-terminal region of the type 2 TGF-β receptor.[54] The exact molecular mechanism is unknown, but as a general overview, DAXX is then phosphorylated by homeodomain-interacting protein kinase 2 (HIPK2), which then activates apoptosis signal-inducing kinase 1 (ASK1), which goes on to activate the Jun amino-terminal kinase (JNK) pathway and thus apoptosis as seen in the left panel of the adjacent image.[55][56]
Effects on immune cells
T lymphocytes
TGF-β1 plays a role in the induction from CD4+ T cells of both induced Tregs (iTregs), which have a regulatory function, and Th17 cells, which secrete pro-inflammatory cytokines.[57][58]
TGF-β1 alone precipitates the expression of Foxp3 and Treg differentiation from activated T helper cells, and the mechanism for this differentiation is unknown for both induced T regulatory cells as well as natural T regulatory cells. In mouse models, the effect of TGF-β1 appears to be age-dependent.[59]
Studies show that neutralization of TGF-β1 in vitro inhibits the differentiation of helper T cells into Th17 cells. The role of TGF-β1 in the generation of Th17 cells goes against its dominant conceptualization as an anti-inflammatory cytokine; however, the shared requirement between inflammatory and anti-inflammatory immune cells suggests that an imbalance between these two cell types can be an important link to autoimmunity.[57] Co-activation by IL-6 from activated dendritic cells, which serves to activate the transcription factor STAT3, is required in addition to TGF-β1 for the differentiation of Th17 cells. However, the molecular mechanism of Th17 differentiation is not well understood.[59] Because Th17 cells are distinct from Th1 and Th2 lineages in that they have been shown to be capable of regulatory functions, this is further evidence of TGF-β1's regulatory function in the immune system.[60]
B lymphocytes
TGF-β has mainly inhibitory effects on B lymphocytes. TGF-β inhibits B cell proliferation. The exact mechanism is unknown, but there is evidence that TGF-β inhibits B cell proliferation by inducing the transcription factor Id3, inducing expression of cyclin-dependent kinase inhibitor 21 (a regulator of cell cycle progression through the G1 and S phase), and repressing other key regulatory genes such as c-myc and ATM.[61][62] CD40, a key surface molecule in the activation of the innate immune response, can induce Smad7 expression to reverse the growth inhibition of B cells induced by TGF-β.[63] TGF-β also blocks B cell activation and promotes class switching IgA in both human and mouse B cells and has an otherwise inhibitory function for antibody production.[61]
TGF-β also induces apoptosis of immature or resting B cells; the mechanism is unknown, but may overlap with its anti-proliferation pathway. TGF-β has been shown to downregulate c-myc as it does in the inhibition of B cell proliferation. It is also known to induce NF-κB inhibitor IKBa, inhibiting NF-κB activation.[64] NF-κB is a transcription factor that regulates the production of cytokines like IL-1, TNF-a, and defensins, although its function in apoptosis may be separate from this function.
Macrophages
The general consensus in the literature is that TGF-β stimulates resting monocytes and inhibits activated macrophages. For monocytes, TGF-β has been shown to function as a chemoattractant as well as an upregulator of inflammatory response.[65] However, TGF-β has also been shown to downregulate inflammatory cytokine production in monocytes and macrophages, likely by the aforementioned inhibition of NF-κB.[66] This contradiction may be due to the fact that the effect of TGF-β has been shown to be highly context-dependent.[67]
TGF-β is thought to play a role in alternative macrophage activation seen in lean mice, and these macrophages maintain an anti-inflammatory phenotype. This phenotype is lost in obese mice, who have not only more macrophages than lean mice but also classically activated macrophages which release TNF-α and other pro-inflammatory cytokines that contribute to a chronically pro-inflammatory milieu.[68]
Cell cycle
TGF-β plays a crucial role in the regulation of the cell cycle by blocking progress through G1 phase. TGF-β causes synthesis of p15 and p21 proteins, which block the cyclin:CDK complex responsible for retinoblastoma protein (Rb) phosphorylation. Thus, TGF-β blocks advancement through the G1 phase of the cycle.[69] In doing so, TGF-β suppresses expression of c-myc, a gene which is involved in G1 cell cycle progression.[69]
Clinical significance
Cancer
In normal cells, TGF-β, acting through its signaling pathway, stops the cell cycle at the G1 stage to stop proliferation, induce differentiation, or promote apoptosis. In many cancer cells, parts of the TGF-β signaling pathway are mutated, and TGF-β no longer controls the cell. These cancer cells proliferate. The surrounding stromal cells (fibroblasts) also proliferate. Both cells increase their production of TGF-β. This TGF-β acts on the surrounding stromal cells, immune cells, endothelial and smooth-muscle cells. It causes immunosuppression and angiogenesis, which makes the cancer more invasive.[70] TGF-β also converts effector T-cells, which normally attack cancer with an inflammatory (immune) reaction, into regulatory (suppressor) T-cells, which turn off the inflammatory reaction. Normal tissue integrity is preserved by feedback interactions between different cell types that express adhesion molecules and secrete cytokines. Disruption of these feedback mechanisms in cancer damages a tissue. When TGF-β signaling fails to control NF-κB activity in cancer cells, this has at least two potential effects: first, it enables the malignant tumor to persist in the presence of activated immune cells, and second, the cancer cell outlasts immune cells because it survives in the presence of apoptotic, and anti-inflammatory mediators.[71]
Heart disease
One animal study suggests that cholesterol suppresses the responsiveness of cardiovascular cells to TGF-β and its protective qualities, thus allowing atherosclerosis and heart disease to develop, while statins, drugs that lower cholesterol levels, may enhance the responsiveness of cardiovascular cells to the protective actions of TGF-β.[72]
TGF-β is involved in regeneration of zebrafish heart.
Marfan syndrome
TGF-β signaling also likely plays a major role in the pathogenesis of Marfan syndrome,[73] a disease characterized by disproportionate height, arachnodactyly, ectopia lentis and heart complications such as mitral valve prolapse and aortic enlargement increasing the likelihood of aortic dissection. While the underlying defect in Marfan syndrome is faulty synthesis of the glycoprotein fibrillin I, normally an important component of elastic fibers, it has been shown that the Marfan syndrome phenotype can be relieved by addition of a TGF-β antagonist in affected mice.[74] This suggests that while the symptoms of Marfan syndrome may seem consistent with a connective tissue disorder, the mechanism is more likely related to reduced sequestration of TGF-β by fibrillin.[75]
Loeys–Dietz syndrome
TGF-β signaling is also disturbed in Loeys–Dietz syndrome which is caused by mutations in the TGF-β receptor.
Obesity and diabetes
TGF-β/SMAD3 signaling pathway is important in regulating glucose and energy homeostasis and might play a role in diabetic nephropathy.
As noted above in the section about macrophages, loss of TGF-β signaling in obesity is one contributor to the inflammatory milieu generated in the case of obesity.[68]
Induced T regulatory cells (iTreg), stimulated by TGF-β in the presence of IL-2, suppressed the development of experimental autoimmune encephalomyelitis (EAE), an animal model of multiple sclerosis (MS) via a Foxp3 and IL-10 mediated response. This suggests a possible role for TGF-β and iTreg in the regulation and treatment of MS.[76]
Decreased levels of TGF-β have been observed in patients diagnosed with multiple sclerosis.[77] Its role in multiple sclerosis can be explained due to TGF-β role in regulating apoptosis of Th17 cells.[77] When TGF-β levels decrease, they are unable to induce Th17 cells apoptosis.[77] Th17 cells secretes TNF-α, which induces demyelination of the oligodendroglial via TNF receptor 1.[78] The decreased TGF-β levels lead to increased Th17 cells and subsequently increased TNFα levels.[77] As a result, demyelination of neurons occurs.[77] TGF-β have also been observed to induce oligodendrocyte (myelin sheath producing cells) growth.[77] Hence, the decreased TGF-β levels during MS may also prevent remyelination of neurons.[77]
Other
Higher concentrations of TGF-β are found in the blood and cerebrospinal fluid of patients with Alzheimer's disease as compared to control subjects,[79] suggesting a possible role in the neurodegenerative cascade leading to Alzheimer's disease symptoms and pathology.
Overactive TGF-β pathway, with an increase of TGF-β2,[80] was reported in the studies of patients suffering from keratoconus.[81]
There is substantial evidence in animal and some human studies that TGF-β in breast milk may be a key immunoregulatory factor in the development of infant immune response, moderating the risk of atopic disease or autoimmunity.[82]
Skin aging is caused in part by TGF-β, which reduces the subcutaneous fat that gives skin a pleasant appearance and texture. TGF-β does this by blocking the conversion of dermal fibroblasts into fat cells; with fewer fat cells underneath to provide support, the skin becomes saggy and wrinkled. Subcutaneous fat also produces cathelicidin, which is a peptide that fights bacterial infections.[83][84]
See also
- Anita Roberts, a molecular biologist who made pioneering observations of TGF-β
References
- doi:10.1038/nrneph.2016.48
- Massagué J (October 2012). "TGFβ signalling in context". Nature Reviews Molecular Cell Biology. 13 (10): 616–30. doi:10.1038/nrm3434. PMC 4027049. PMID 22992590.
- Nakao A, Afrakhte M, Morén A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P (October 1997). "Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling". Nature. 389 (6651): 631–5. doi:10.1038/39369. PMID 9335507.
- AfCS signaling gateway - data center - ligand description
- Letterio JJ, Roberts AB (April 1998). "Regulation of immune responses by TGF-beta". Annual Review of Immunology. 16 (1): 137–61. doi:10.1146/annurev.immunol.16.1.137. PMID 9597127.
- Massagué J, Xi Q (July 2012). "TGF-β control of stem cell differentiation genes". FEBS Letters. 586 (14): 1953–8. doi:10.1016/j.febslet.2012.03.023. PMC 3466472. PMID 22710171.
- Li MO, Flavell RA (August 2008). "TGF-beta: a master of all T cell trades". Cell. 134 (3): 392–404. doi:10.1016/j.cell.2008.07.025. PMC 3677783. PMID 18692464.
- Massagué J, Blain SW, Lo RS (October 2000). "TGFbeta signaling in growth control, cancer, and heritable disorders". Cell. 103 (2): 295–309. doi:10.1016/S0092-8674(00)00121-5. PMID 11057902.
- Online Mendelian Inheritance in Man (OMIM): 190180
- Universal protein resource accession number P01137 at UniProt.
- Online Mendelian Inheritance in Man (OMIM): 190220
- Universal protein resource accession number P61812 at UniProt.
- Online Mendelian Inheritance in Man (OMIM): 190230
- Universal protein resource accession number P10600 at UniProt.
- Roberts AB, Kim SJ, Noma T, Glick AB, Lafyatis R, Lechleider R, Jakowlew SB, Geiser A, O'Reilly MA, Danielpour D (1991). "Multiple forms of TGF-beta: distinct promoters and differential expression". Ciba Found Symp. Novartis Foundation Symposia. 157: 7–15, discussion 15–28. doi:10.1002/9780470514061.ch2. ISBN 9780470514061. PMID 1906395.
- Jugessur A, Shi M, Gjessing HK, et al. (2010). "Maternal genes and facial clefts in offspring: a comprehensive search for genetic associations in two population-based cleft studies from Scandinavia". PLOS ONE. 5 (7): e11493. doi:10.1371/journal.pone.0011493. PMC 2901336. PMID 20634891.
- He C, Kraft P, Chasman DI, et al. (November 2010). "A large-scale candidate gene association study of age at menarche and age at natural menopause". Hum. Genet. 128 (5): 515–27. doi:10.1007/s00439-010-0878-4. PMC 2967297. PMID 20734064.
- Otsuki T, Ota T, Nishikawa T, et al. (2005). "Signal sequence and keyword trap in silico for selection of full-length human cDNAs encoding secretion or membrane proteins from oligo-capped cDNA libraries". DNA Res. 12 (2): 117–26. doi:10.1093/dnares/12.2.117. PMID 16303743.
- Cornet PB, Galant C, Eeckhout Y, Courtoy PJ, Marbaix E, Henriet P (2005). "Regulation of matrix metalloproteinase-9/gelatinase B expression and activation by ovarian steroids and LEFTY-A/endometrial bleeding-associated factor in the human endometrium". J Clin Endocrinol Metab. 90 (2): 1001–11. doi:10.1210/jc.2004-1277. PMID 15536155.
- Tabibzadeh S, Kothapalli R, Buyuksal I (1997). "Distinct tumor specific expression of TGFB4 (ebaf)*, a novel human gene of the TGF-beta superfamily". Front. Biosci. 2: a18–25. doi:10.2741/a158. PMID 9230066.
- Khalil N (1999). "TGF-beta: from latent to active". Microbes Infect. 1 (15): 1255–63. doi:10.1016/S1286-4579(99)00259-2. PMID 10611753.
- Herpin A, Lelong C, Favrel P (2004). "Transforming growth factor-beta-related proteins: an ancestral and widespread superfamily of cytokines in metazoans". Dev Comp Immunol. 28 (5): 461–85. doi:10.1016/j.dci.2003.09.007. PMID 15062644.
- Daopin S, Piez K, Ogawa Y, Davies D (1992). "Crystal structure of transforming growth factor-beta 2: an unusual fold for the superfamily". Science. 257 (5068): 369–73. doi:10.1126/science.1631557. PMID 1631557.
- Derynck R, Jarrett JA, Chen EY, et al. (1985). "Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells". Nature. 316 (6030): 701–5. doi:10.1038/316701a0. PMID 3861940.
- Rifkin DB (March 2005). "Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability". J. Biol. Chem. 280 (9): 7409–12. doi:10.1074/jbc.R400029200. PMID 15611103.
- Dubois CM, Laprise MH, Blanchette F, Gentry LE, Leduc R (May 1995). "Processing of transforming growth factor beta 1 precursor by human furin convertase". J. Biol. Chem. 270 (18): 10618–24. doi:10.1074/jbc.270.18.10618. PMID 7737999.
- Annes JP, Munger JS, Rifkin DB (January 2003). "Making sense of latent TGFbeta activation". J. Cell Sci. 116 (Pt 2): 217–24. doi:10.1242/jcs.00229. PMID 12482908.
- Saharinen J, Hyytiäinen M, Taipale J, Keski-Oja J (June 1999). "Latent transforming growth factor-beta binding proteins (LTBPs)--structural extracellular matrix proteins for targeting TGF-beta action". Cytokine Growth Factor Rev. 10 (2): 99–117. doi:10.1016/S1359-6101(99)00010-6. PMID 10743502.
- Sterner-Kock A, Thorey IS, Koli K, et al. (September 2002). "Disruption of the gene encoding the latent transforming growth factor-beta binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer". Genes Dev. 16 (17): 2264–73. doi:10.1101/gad.229102. PMC 186672. PMID 12208849.
- Saharinen J, Keski-Oja J (August 2000). "Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta". Mol. Biol. Cell. 11 (8): 2691–704. doi:10.1091/mbc.11.8.2691. PMC 14949. PMID 10930463.
- ten Dijke P, Hill CS (May 2004). "New insights into TGF-beta-Smad signalling". Trends Biochem. Sci. 29 (5): 265–73. doi:10.1016/j.tibs.2004.03.008. PMID 15130563.
- Stetler-Stevenson WG, Aznavoorian S, Liotta LA (1993). "Tumor cell interactions with the extracellular matrix during invasion and metastasis". Annu. Rev. Cell Biol. 9: 541–73. doi:10.1146/annurev.cb.09.110193.002545. PMID 8280471.
- Barcellos-Hoff MH, Dix TA (September 1996). "Redox-mediated activation of latent transforming growth factor-beta 1". Mol. Endocrinol. 10 (9): 1077–83. doi:10.1210/me.10.9.1077. PMID 8885242.
- Wipff PJ, Hinz B (September 2008). "Integrins and the activation of latent transforming growth factor beta1 — an intimate relationship". Eur. J. Cell Biol. 87 (8–9): 601–15. doi:10.1016/j.ejcb.2008.01.012. PMID 18342983.
- Yu Q, Stamenkovic I (January 2000). "Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis". Genes Dev. 14 (2): 163–76. PMC 316345. PMID 10652271.
- Taipale J, Miyazono K, Heldin CH, Keski-Oja J (January 1994). "Latent transforming growth factor-beta 1 associates to fibroblast extracellular matrix via latent TGF-beta binding protein". J. Cell Biol. 124 (1–2): 171–81. doi:10.1083/jcb.124.1.171. PMC 2119892. PMID 8294500.
- Lyons RM, Keski-Oja J, Moses HL (May 1988). "Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium". J. Cell Biol. 106 (5): 1659–65. doi:10.1083/jcb.106.5.1659. PMC 2115066. PMID 2967299.
- Booth WJ, Berndt MC (July 1987). "Thrombospondin in clinical disease states". Semin. Thromb. Hemost. 13 (3): 298–306. doi:10.1055/s-2007-1003505. PMID 3317840.
- Raugi GJ, Olerud JE, Gown AM (December 1987). "Thrombospondin in early human wound tissue". J. Invest. Dermatol. 89 (6): 551–4. doi:10.1111/1523-1747.ep12461198. PMID 3680981.
- Schultz-Cherry S, Murphy-Ullrich JE (August 1993). "Thrombospondin causes activation of latent transforming growth factor-beta secreted by endothelial cells by a novel mechanism". J. Cell Biol. 122 (4): 923–32. doi:10.1083/jcb.122.4.923. PMC 2119591. PMID 8349738.
- Murphy-Ullrich JE, Poczatek M (2000). "Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology". Cytokine Growth Factor Rev. 11 (1–2): 59–69. doi:10.1016/S1359-6101(99)00029-5. PMID 10708953.
- Huang XZ, Wu JF, Cass D, et al. (May 1996). "Inactivation of the integrin beta 6 subunit gene reveals a role of epithelial integrins in regulating inflammation in the lung and skin". J. Cell Biol. 133 (4): 921–8. doi:10.1083/jcb.133.4.921. PMC 2120829. PMID 8666675.
- Bader BL, Rayburn H, Crowley D, Hynes RO (November 1998). "Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins". Cell. 95 (4): 507–19. doi:10.1016/S0092-8674(00)81618-9. PMID 9827803.
- Shull MM, Ormsby I, Kier AB, et al. (October 1992). "Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease". Nature. 359 (6397): 693–9. doi:10.1038/359693a0. PMC 3889166. PMID 1436033.
- Munger JS, Harpel JG, Giancotti FG, Rifkin DB (September 1998). "Interactions between growth factors and integrins: latent forms of transforming growth factor-beta are ligands for the integrin alphavbeta1". Mol. Biol. Cell. 9 (9): 2627–38. CiteSeerX 10.1.1.492.5809. doi:10.1091/mbc.9.9.2627. PMC 25536. PMID 9725916.
- Munger JS, Huang X, Kawakatsu H, et al. (February 1999). "The integrin alpha v beta 6 binds and activates latent TGF beta 1: a mechanism for regulating pulmonary inflammation and fibrosis". Cell. 96 (3): 319–28. doi:10.1016/S0092-8674(00)80545-0. PMID 10025398.
- Kulkarni AB, Huh CG, Becker D, et al. (January 1993). "Transforming growth factor β 1 null mutation in mice causes excessive inflammatory response and early death". Proc. Natl. Acad. Sci. U.S.A. 90 (2): 770–4. doi:10.1073/pnas.90.2.770. PMC 45747. PMID 8421714.
- Taylor AW (January 2009). "Review of the activation of TGF-beta in immunity". J. Leukoc. Biol. 85 (1): 29–33. doi:10.1189/jlb.0708415. PMC 3188956. PMID 18818372.
- Mu D, Cambier S, Fjellbirkeland L, et al. (April 2002). "The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1". J. Cell Biol. 157 (3): 493–507. doi:10.1083/jcb.200109100. PMC 2173277. PMID 11970960.
- Derynck, Rik; Zhang, Ying; Feng, Xin-Hua (1998-12-11). "Transcriptional Activators of TGF-β Responses: Smads". Cell. 95 (6): 737–740. doi:10.1016/S0092-8674(00)81696-7. ISSN 0092-8674. PMID 9865691.
- Derynck, Rik; Zhang, Ying E. (October 2003). "Smad-dependent and Smad-independent pathways in TGF-β family signalling". Nature. 425 (6958): 577–584. doi:10.1038/nature02006. PMID 14534577.
- Letterio, John J. (2005-01-01). "TGF-β signaling in T cells: roles in lymphoid and epithelial neoplasia". Oncogene. 24 (37): 5701–5712. doi:10.1038/sj.onc.1208922. ISSN 0950-9232. PMID 16123803.
- Kulkarni, A. B.; Huh, C. G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K. C.; Roberts, A. B.; Sporn, M. B.; Ward, J. M. (1993). "Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death". Proceedings of the National Academy of Sciences. 90 (2): 770–774. doi:10.1073/pnas.90.2.770. ISSN 0027-8424. PMC 45747. PMID 8421714.
- Yang, Xiaolu; Khosravi-Far, Roya; Chang, Howard Y.; Baltimore, David (1997-06-27). "Daxx, a Novel Fas-Binding Protein That Activates JNK and Apoptosis". Cell. 89 (7): 1067–1076. doi:10.1016/S0092-8674(00)80294-9. ISSN 0092-8674. PMC 2989411. PMID 9215629.
- Perlman, Riki; Schiemann, William P.; Brooks, Mary W.; Lodish, Harvey F.; Weinberg, Robert A. (August 2001). "TGF-β-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation". Nature Cell Biology. 3 (8): 708–714. doi:10.1038/35087019. PMID 11483955.
- Hofmann, Thomas G.; Stollberg, Nicole; Schmitz, M. Lienhard; Will, Hans (2003-12-01). "HIPK2 regulates transforming growth factor-beta-induced c-Jun NH(2)-terminal kinase activation and apoptosis in human hepatoma cells". Cancer Research. 63 (23): 8271–8277. ISSN 0008-5472. PMID 14678985.
- Eisenstein, Eli M.; Williams, Calvin B. (2009-05-01). "The Treg/Th17 Cell Balance: A New Paradigm for Autoimmunity". Pediatric Research. 65 (5 Part 2): 26R–31R. doi:10.1203/PDR.0b013e31819e76c7. ISSN 0031-3998. PMID 19218879.
- Morishima, Noriko; Mizoguchi, Izuru; Takeda, Kiyoshi; Mizuguchi, Junichiro; Yoshimoto, Takayuki (2009-08-14). "TGF-β is necessary for induction of IL-23R and Th17 differentiation by IL-6 and IL-23". Biochemical and Biophysical Research Communications. 386 (1): 105–110. doi:10.1016/j.bbrc.2009.05.140. PMID 19501566.
- Li, Ming O.; Flavell, Richard A. (2008-08-08). "TGF-β: A Master of All T Cell Trades". Cell. 134 (3): 392–404. doi:10.1016/j.cell.2008.07.025. ISSN 0092-8674. PMC 3677783. PMID 18692464.
- O'Connor, William; Zenewicz, Lauren A; Flavell, Richard A (2010). "The dual nature of TH17 cells: shifting the focus to function". Nature Immunology. 11 (6): 471–476. doi:10.1038/ni.1882. PMID 20485275.
- Li, Ming O.; Wan, Yisong Y.; Sanjabi, Shomyseh; Robertson, Anna-Karin L.; Flavell, Richard A. (2006-01-01). "TRANSFORMING GROWTH FACTOR-β REGULATION OF IMMUNE RESPONSES". Annual Review of Immunology. 24 (1): 99–146. doi:10.1146/annurev.immunol.24.021605.090737. PMID 16551245.
- Roes, Jürgen; Choi, B. Ken; Cazac, Balthazar B. (2003-06-10). "Redirection of B cell responsiveness by transforming growth factor β receptor". Proceedings of the National Academy of Sciences. 100 (12): 7241–7246. doi:10.1073/pnas.0731875100. ISSN 0027-8424. PMC 165860. PMID 12773615.
- Patil, Supriya; Wildey, Gary M.; Brown, Thomas L.; Choy, Lisa; Derynck, Rik; Howe, Philip H. (2000-12-08). "Smad7 Is Induced by CD40 and Protects WEHI 231 B-lymphocytes from Transforming Growth Factor-β-induced Growth Inhibition and Apoptosis". Journal of Biological Chemistry. 275 (49): 38363–38370. doi:10.1074/jbc.M004861200. ISSN 0021-9258. PMID 10995749.
- Arsura, Marcello; Wu, Min; Sonenshein, Gail E. (1996-07-01). "TGFβ1 Inhibits NF-κB/Rel Activity Inducing Apoptosis of B Cells: Transcriptional Activation of IκBα". Immunity. 5 (1): 31–40. doi:10.1016/S1074-7613(00)80307-6. ISSN 1074-7613. PMID 8758892.
- Kubiczkova, Lenka; Sedlarikova, Lenka; Hajek, Roman; Sevcikova, Sabina (2012-09-03). "TGF-β – an excellent servant but a bad master". Journal of Translational Medicine. 10 (1): 183. doi:10.1186/1479-5876-10-183. PMC 3494542. PMID 22943793.
- Smythies, Lesley E.; Sellers, Marty; Clements, Ronald H.; Mosteller-Barnum, Meg; Meng, Gang; Benjamin, William H.; Orenstein, Jan M.; Smith, Phillip D. (2005-01-03). "Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity". Journal of Clinical Investigation. 115 (1): 66–75. doi:10.1172/JCI19229. PMC 539188. PMID 15630445.
- Wahl, Sharon M (February 2007). "Transforming growth factor-β: innately bipolar". Current Opinion in Immunology. 19 (1): 55–62. doi:10.1016/j.coi.2006.11.008. PMID 17137775.
- Wu, Davina; Molofsky, Ari B.; Liang, Hong-Erh; Ricardo-Gonzalez, Roberto R.; Jouihan, Hani A.; Bando, Jennifer K.; Chawla, Ajay; Locksley, Richard M. (2011-04-08). "Eosinophils Sustain Adipose Alternatively Activated Macrophages Associated with Glucose Homeostasis". Science. 332 (6026): 243–247. doi:10.1126/science.1201475. ISSN 0036-8075. PMC 3144160. PMID 21436399.
- Hanahan D, Weinberg RA (January 2000). "The hallmarks of cancer". Cell. 100 (1): 57–70. doi:10.1016/S0092-8674(00)81683-9. PMID 10647931.
- Blobe GC, Schiemann WP, Lodish HF (May 2000). "Role of transforming growth factor beta in human disease". N. Engl. J. Med. 342 (18): 1350–8. doi:10.1056/NEJM200005043421807. PMID 10793168.
- Vlahopoulos, SA; Cen, O; Hengen, N; Agan, J; Moschovi, M; Critselis, E; Adamaki, M; Bacopoulou, F; Copland, JA; Boldogh, I; Karin, M; Chrousos, GP (20 June 2015). "Dynamic aberrant NF-κB spurs tumorigenesis: A new model encompassing the microenvironment". Cytokine & Growth Factor Reviews. 26 (4): 389–403. doi:10.1016/j.cytogfr.2015.06.001. PMC 4526340. PMID 26119834.
- Understanding Heart Disease: Research Explains Link Between Cholesterol and Heart Disease Archived 2007-11-12 at the Wayback Machine
- Entrez Gene (2007). "TGFBR2 transforming growth factor, beta receptor II" (Entrez gene entry). Retrieved January 11, 2007.
- Habashi JP, Judge DP, Holm TM, et al. (April 2006). "Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome". Science. 312 (5770): 117–21. doi:10.1126/science.1124287. PMC 1482474. PMID 16601194.
- Robinson PN, Arteaga-Solis E, Baldock C, et al. (October 2006). "The molecular genetics of Marfan syndrome and related disorders". J. Med. Genet. 43 (10): 769–87. doi:10.1136/jmg.2005.039669. PMC 2563177. PMID 16571647.
- Selvaraj RK, Geiger T (March 2008). "Mitigation of experimental allergic encephalomyelitis by TGF-beta induced Foxp3+ regulatory T lymphocytes through the induction of anergy and infectious tolerance". Journal of Immunology. 180 (5): 2830–2838. doi:10.4049/jimmunol.180.5.2830. PMID 18292504.
- Dobolyi A, Vincze C, Pál G, Lovas G (July 2012). "The neuroprotective functions of transforming growth factor Beta proteins". Int J Mol Sci. 13 (7): 8219–58. doi:10.3390/ijms13078219. PMC 3430231. PMID 22942700.
- Nakahara J, Maeda M, Aiso S, Suzuki N (February 2012). "Current concepts in multiple sclerosis: autoimmunity versus oligodendrogliopathy". Clinical Reviews in Allergy & Immunology. 42 (1): 26–34. doi:10.1007/s12016-011-8287-6. PMID 22189514.
- Swardfager W, Lanctôt K, Rothenburg L, Wong A, Cappell J, Herrmann N (November 2010). "A meta-analysis of cytokines in Alzheimer's disease". Biol. Psychiatry. 68 (10): 930–41. doi:10.1016/j.biopsych.2010.06.012. PMID 20692646.
- Maier P, Broszinski A, Heizmann U, Böhringer D, Reinhardau T (2007). "Active transforming growth factor-beta2 is increased in the aqueous humor of keratoconus patients". Molecular Vision. 13: 1198–202. PMID 17679942.
- Engler C, Chakravarti S, Doyle J, Eberhart CG, Meng H, Stark WJ, Kelliher C, Jun AS (May 2011). "Transforming growth factor-β signaling pathway activation in Keratoconus". American Journal of Ophthalmology. 151 (5): 752–759.e2. doi:10.1016/j.ajo.2010.11.008. PMC 3079764. PMID 21310385.
- Oddy, Wendy H.; Rosales, Francisco (2010-02-01). "A systematic review of the importance of milk TGF-β on immunological outcomes in the infant and young child". Pediatric Allergy and Immunology. 21 (1–Part–I): 47–59. doi:10.1111/j.1399-3038.2009.00913.x. ISSN 1399-3038. PMID 19594862.
- Galindo, Yadira (2018-12-26). "UC San Diego Researchers Identify How Skin Ages, Loses Fat and Immunity" (Press release). University of California San Diego.
- Zhang, Ling-juan & 13 co-authors (2019). "Age-Related Loss of Innate Immune Antimicrobial Function Of Dermal Fat Is Mediated By Transforming Growth Factor Beta". Immunity. 50 (1): 121–136.e5. doi:10.1016/j.immuni.2018.11.003. PMID 30594464.
Further reading
- Moses HL, Roberts AB, Derynck R (2016). "The Discovery and Early Days of TGF-β: A Historical Perspective". Cold Spring Harbor Perspectives in Biology. 8 (7): a021865. doi:10.1101/cshperspect.a021865. PMC 4930926. PMID 27328871.
External links
- Description of the TGF beta producing genes at ncbi.nlm.nih.gov
- Diagram of the TGF beta signaling pathway at genome.ad.jp
- The TGF-beta system — Nature Reviews Molecular Cell Biology
- SMART:TGFB domain annotation — European Molecular Biology Laboratory Heidelberg
- TGF-beta at the US National Library of Medicine Medical Subject Headings (MeSH)
- Biochemists Solve Structure Of TGF-Beta And Its Receptor. 2008 – shows TGF-β3 dimer in TGFB-receptor
- Measurement of Human Latent TGF-β1
- TGF beta pathway diagram
- IntroPro Entry - IPR016319