RNase MRP
RNase MRP (also called RMRP) is an enzymatically active ribonucleoprotein with two distinct roles in eukaryotes. RNAse MRP stands for RNAse for mitochondrial RNA processing. In mitochondria it plays a direct role in the initiation of mitochondrial DNA replication. In the nucleus it is involved in precursor rRNA processing, where it cleaves the internal transcribed spacer 1 between 18S and 5.8S rRNAs.[1] Despite distinct functions, RNase MRP has been shown to be evolutionarily related to RNase P. Like eukaryotic RNase P, RNase MRP is not catalytically active without associated protein subunits.[2]
| RNase MRP | |
|---|---|
 Predicted secondary structure and sequence conservation of RNase_MRP | |
| Identifiers | |
| Symbol | RNase MRP |
| Rfam | RF00030 |
| Other data | |
| RNA type | Gene; ribozyme |
| Domain(s) | Eukaryota |
| GO | 0006364 0000171 0000172 |
| SO | 0000385 |
| PDB structures | PDBe |
Mutations in the RNA component of RNase MRP cause cartilage–hair hypoplasia, a pleiotropic human disease. Responsible for this disease is a mutation in the RNase MRP RNA gene (RMRP), a non-coding RNA gene. RMRP was the first non-coding nuclear RNA gene found to cause disease.[3]
Mechanism and mutation effects

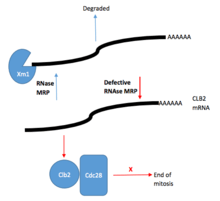
RNAase MRP and its role in pre-rRNA processing has been previously studied in Yeast cells. RNase MRP has been shown to cleave an internal transcribed spacer, specifically ITS1 at the specific site A3 of the rRNA precursor, leading, after additional trimming, to the formation of the mature 5′-end of 5.8S rRNA. Recent data that has been gathered using several temperature-sensitive RNase MRP mutants that showed that inactivation of RNase MRP leading to severe reduction of the abundance of all early intermediates in the typical rRNA processing pathway. However, the transcription of the rRNA precursor is not affected, thus suggesting that RNase MRP plays a key role in the processing of rRNA beyond the cleavage of the A3 site in ITS1. Further research in Yeast cell RNase MRP has shown a potential role in the regulation of the cell cycle. RNase MRP mutations led to missegregation of plasmids and caused cell cycle delay at the end of mitosis, followed by a buildup of cyclin B2 (CLB2) protein (resulting from increased CLB2 mRNA concentration that codes for the CLB2 protein). RNase MRP also demonstrated cleavage ability of the 5′-UTR of CLB2 mRNA that allows for rapid 5′-to-3′ degradation by XRN1, an exoribonuclease enzyme.[4]
Link to RNAse P
RNase P and RNAse MRP are ribonucleoprotein complexes that are important in RNA processing. Both subunits have a highly conserved P4 helical region, which is a type of nucleic acid tertiary structure. This region is needed for catalytic function, and is probably an important part of the enzyme's active site. RNAse P is found in both eukaryotes and prokaryotes and it cleaves a pre-tRNA to generate the mature 5’ end of the tRNA. RNase MRP is found only in eukaryotes, and is involved in rRNA processing, which is the conversion of preribosomal RNA into mature rRNA through splicing, modifications, and cleavage. The exact mechanism is described above.[5]
Evolutionary link
.png)
These two ribonucleases are most likely evolutionarily related through a common ancestor, since they have common protein subunits and can be folded into very similar secondary structures. There are many conserved regions in these two ribonucleases. Sequences of the CR-I, CR-V, and CR-IV genes in domain 1 of the P4 helical region are conserved, with the consensus sequence in CR-IV being AGNNNNA for RNAse P and AGNNA for RNase MRP. CR-II and CR-III are also conserved in domain 2 of P RNA. The P3 helix is also conserved in both ribonucleases in all eukaryotes, but the function of this helix is not yet clear. These conserved regions are evidence of the close phylogenetic relation between these two important ribonucleoprotein complexes.[5]
Diseases associated with RNAse MRP gene
Metaphyseal dysplasia without hypotrichosis (MDWH), anauxetic dysplasia (AD), kyphomelic dysplasia (KD), Omenn syndrome (OS) are diseases associated with mutated and (or) dysfunctional RNAse MRP activity, hence, the RMRP gene.
| Disease | Abbreviation | Location of mutation | Mutation in RNAse MRP protein or RNA in RNAse MRP? | Symptoms |
|---|---|---|---|---|
| Cartilage–hair hypoplasia | CHH | 1.Insertion, duplication or triplication at promoter or 2. In RNA transcribed by RNAse MRP | RNA in RNAse MRP | Patients have short stature, skeletal anomalies, blood and immune problems, and fine, light-colored hair |
| Metaphyseal dysplasia without hypotrichosis | MDWH | 1. on RMRP Gene-->common insertion being -21-20insTCTGTGAAGCTGGGGAC on paternal allele or 2. 218A-->G point mutation occurring on maternal allele | RNA in RNAse MRP | Patients unable to produce new tubular structures in metaphyses of long bones. This results in porous and expanded long bones |
| Anauxetic dysplasia | AD | Homozygous insertion mutation and two compound heterozygous mutations | RNA in RNAse MRP | Early onset of extremely short stature. Adults typically not exceeding 85 cm in height. Abnormal amount of teeth (less than standard amount). Slight mental retardation. |
| Kyphomelic dysplasia | KD | Mutation (insertion) of T at 194-195 paternal allele and a 63 C-->T point mutation of the maternal allele. | Undetermined | Form of short-limbed dwarfism. Bowing long bones, dysmorphia, flattened vertebrae, and short ribs. |
| Omenn Syndrome | OS | Three mutations in RMRP gene (specifics unknown at this time) | RNA in RNAse MRP | Patients are immunodeficient and have scaly erythroderma and severe reddening of skin. |
Cartilage–hair hypoplasia
Mutations in the RNA component of RNase MRP cause cartilage–hair hypoplasia(CHH), a pleiotropic human disease. Two categories of mutations involving RNAse MRP have been identified in patients with CHH. The first type is when an insertion, duplication, or triplication occurs at the promoter of the RNAse MRP gene between the TATA box and the transcription initiation site. This causes the initiation of RNAse MRP to be slow, or to not occur at all. The second category consists of mutations that are in the transcribed RNA made by the RNAse MRP. Patients with CHH have been identified to have over 70 different mutations in the RNA transcript made by RNAse MRP, whereas around 30 distinct mutations have been identified in the promoter region of the RNAse MRP gene. Most CHH patients have a combination of either a promoter mutation in one allele along with a RNAse MRP RNA mutation in the other allele, or a combination of two RNAse MRP RNA mutations in both alleles. The fact that there is not often a mutation in the promoter region in both alleles shows the lethality of not having this RNA present that is transcribed by RNAse MRP.[6][7][8]
Metaphyseal dysplasia without hypotrichosis
Metaphyseal dysplasia Without Hypotrichosis (MDWH) patients are unable to produce normal, new tubular structures in the metaphyses of long bones. People diagnosed with MDWH will therefore tend to experience porous and expanded long bones. The mutation occurs on the RMRP gene in MDWH; the common insertion being (-21-20 insTCTGTGAAGCTGGGGAC) on the paternal allele and a 218A→G point mutation occurring on the maternal allele. MDWH is most likely a variant of CHH. They are the same in that they both display short stature. Some of the same genes involved in the mutations in CHH are the same genes that are mutated in MDWH.[9] These two diseases do differ in that MDWH lacks immunodeficiency and other skeletal features found in CHH patients.[3]
Anauxetic dysplasia
AD is an autosomal recessive spondylometaepiphyseal dysplasia typically characterized by an early (prenatal) onset of extremely short stature and adults that do not typically exceed 85 cm in height. A less than normal amount of teeth and slight mental retardation are also typical of AD. The associated mutation(s) are a homozygous insertion mutation and two compound heterozygous mutations.[3] Mutations in the promoter 5' regulatory region have been associated with this severe skeletal disease. Other names used to describe this condition are spondylometaepiphyseal dysplasia, anauxetic type, spondylometaepiphyseal dysplasia, Menger type.[10]
Kyphomelic dysplasia
KD is a form of short-limbed dwarfism. Characteristics of KD are bowing of long bones, dysmorphia, flattened vertebrae, and short ribs. Femoral bowing is the hallmark diagnostic characteristic of KD. Novel mutations have been discovered in the RMRP gene of a single patient with KD, specifically, a mutation (insertion) of T at 194-195 paternal allele and a 63C-->T point mutation of the maternal allele. As with OS, the MSRP gene has not been strictly linked to the diseases but current research is suggestive that the MSRP gene is a factor. KD has been observed in very few patients yet this sublethal disease remains relevant to discussions of the distinct manifestations of minimal change disease. KD is rather similar to several forms of MCD in that it exhibits combined immune deficiency and aplastic anemia.[3]
Omenn syndrome
Omenn syndrome (OS) is a severe immunodeficiency disease, mostly characterized by scaly erythroderma and severe reddening of the skin. OS is also commonly accompanied by enlarged lymphoid tissues, protracted diarrhea, failure to thrive, and eosinophilia. Gene sequences of people with OS reveal three novel mutations in the RMRP gene, suggesting a link to the RMRP gene, but research is ongoing to better ascertain the cause of OS. At the moment there exists only one treatment for OS which is bone marrow transplantation. If no treatment is performed OS is rather fatal resulting in death in infancy. Patients with OS are immunodeficient meaning their immune system is compromised and cannot properly fight infections resulting in serious secondary illnesses.[3]
References
- Li X, Frank DN, Pace N, Zengel JM, Lindahl L (June 2002). "Phylogenetic analysis of the structure of RNase MRP RNA in yeasts". RNA. 8 (6): 740–51. doi:10.1017/S1355838202022082. PMC 1370293. PMID 12088147.
- Kiss T, Marshallsay C, Filipowicz W (October 1992). "7-2/MRP RNAs in plant and mammalian cells: association with higher order structures in the nucleolus". The EMBO Journal. 11 (10): 3737–46. doi:10.1002/j.1460-2075.1992.tb05459.x. PMC 556834. PMID 1382978.
- Martin AN, Li Y (March 2007). "RNase MRP RNA and human genetic diseases". Cell Research. 17 (3): 219–26. doi:10.1038/sj.cr.7310120. PMID 17189938.
- Esakova O, Krasilnikov AS (September 2010). "Of proteins and RNA: the RNase P/MRP family". RNA. 16 (9): 1725–47. doi:10.1261/rna.2214510. PMC 2924533. PMID 20627997.
- Piccinelli P, Rosenblad MA, Samuelsson T (July 21, 2005). "Identification and analysis of ribonuclease P and MRP RNA in a broad range of eukaryotes". Nucleic Acids Research. 33 (14): 4485–95. doi:10.1093/nar/gki756. PMC 1183490. PMID 16087735.
- Integrated Genetics (2015). "Cartilage-hair Hypoplasia". Integrated Genetics. Laboratory Corporation of America. Retrieved 10 November 2015.
- Mattijssen S, Welting TJ, Pruijn GJ (2010). "RNase MRP and disease". Wiley Interdisciplinary Reviews: RNA. 1 (1): 102–16. doi:10.1002/wrna.9. PMID 21956908.
- Bradshaw, Ralph; Stahl, Phillip (2015). Encyclopedia of Cell Biology. Academic Press. pp. 294–295. ISBN 9780123947963.
- U.S. National Library of Medicine. "RMRP". Genetics Home Reference. Retrieved 12 November 2015.
- HealthGrades. "What is Anauxetic Dysplasia?". Right Diagnosis. Retrieved 13 November 2015.
External links
- Page for RNase MRP at Rfam
- RNase+MRP at the US National Library of Medicine Medical Subject Headings (MeSH)