Adenylosuccinate lyase deficiency
Adenylosuccinate lyase deficiency, is a rare autosomal recessive metabolic disorder characterized by the appearance of succinylaminoimidazolecarboxamide riboside (SAICA riboside) and succinyladenosine (S-Ado) in cerebrospinal fluid, urine.[5][6] These two succinylpurines are the dephosphorylated derivatives of SAICA ribotide (SAICAR) and adenylosuccinate (S-AMP), the two substrates of adenylosuccinate lyase (ADSL), which catalyzes an important reaction in the de novo pathway of purine biosynthesis. ADSL catalyzes two distinct reactions in the synthesis of purine nucleotides, both of which involve the β-elimination of fumarate to produce aminoimidazole carboxamide ribotide (AICAR) from SAICAR or adenosine monophosphate (AMP) from S-AMP.[6][5][4]
| Adenylosuccinate lyase deficiency | |
|---|---|
| Other names | Adenylosuccinase deficiency,[1] |
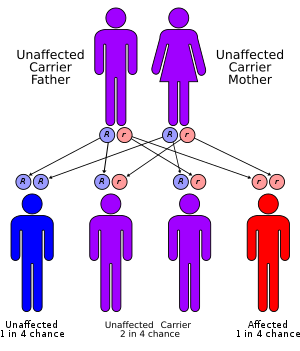 | |
| Adenylosuccinate lyase deficiency has an autosomal recessive pattern of inheritance | |
| Symptoms | Aggressive behavior, Microcephaly[2] |
| Causes | Lack of the enzyme adenylosuccinate lyase[2] |
| Diagnostic method | MRI, Genetic testing[3][4] |
| Treatment | D-ribose and uridine administration[5] |
Presentation
Among the signs and symptoms of adenylosuccinate lyase deficiency are the following:[2]
- Aggressive behavior
- Microcephaly
- Autism
- Brachycephaly
- Mild Cerebellar hypoplasia
- Seizures
Pathophysiology
Adenylosuccinate lyase deficiency is responsible for a range of symptoms that involve psychomotor retardation, often accompanied by epileptic seizures, and autistic features. Two common theories were proposed to account for these effects, the first is that they result from decreased concentrations of purine nucleotides needed for purine biosynthesis. Decreased concentrations, however, could not be found in various tissues taken from ADSL-deficient people, probably because purines are furnished via the purine salvage pathway.[7] The second is the buildup of accumulating succinylpurines causes neurotoxic effects. In the severely affected individuals, the concentration levels of SAICA riboside and S-Ado are comparable, whereas in people with milder forms of the disease, the ratio of S-Ado is more than double that of those more severely affected, while SAICA riboside concentration levels remain comparable.

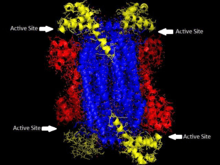
Biochemical studies of the enzyme have focused on proteins of ADSL from nonhuman species, the ADSL structure from the crystallized protein of Thermotoga maritime has been used, along with DNA sequencing data, to construct homology models for a variety of other organisms, including human ADSL. A variety of studies have been done using the equivalent enzyme from Bacillus subtilis, which shares a significant percentage of identity along with about some percentage of similarity in amino acid sequence with the human enzyme. Homology models overlaid on each other show a high degree of overlap between the enzymes. [8]
The family of enzymes to which ADSL belongs and that catalyze β-eliminations in which fumarate is one of the products are homotetramers with four active sites composed of amino acid residues from three distinct subunits. Much is known about the active site of human ADSL due to studies of the active site in the B. subtilis ADSL through affinity labeling and site-directed mutagenesis. While there is some variability among species in the sequencing of ADSL, the active site of the enzyme contains many residues that are conserved across species and have been shown to be critical to the enzyme's function. His68 and His141 seem to serve as the general acid and base catalysts, and are critical to the catalyzing reaction of the substrate. His89 seems to enhance the binding of the substrate's phosphoryl group and orient adenylosuccinate for catalysis. All three histidines are conserved throughout the 28 species for which the structure of ADSL is known. Glu275 and Lys268 have also been shown to contribute to the active site, indicating there are four active sites, each of which is formed from regions of three subunits. [8]ADSL deficiency in different people is often caused by different mutations to the enzyme, more than 50 different mutations in the ADSL gene have been discovered[9]
Diagnosis
In terms of the diagnosis of adenylosuccinate lyase deficiency one should look for (or exam/method):[4][3]
- MRI
- Demonstration of succinylpurines in extracellular fluids like plasma, cerebrospinal fluid and/or urine using high-pressure liquid chromatography, with or without mass spectroscopy
- Genetic testing - genomic cDNA sequencing of the ADSL gene and characterization of mutant proteins.
Treatment
Treatment of adenylosuccinate lyase deficiency can be done via epilepsy management with anticonvulsive drugs. Additionally the following options include:[5]
- D-ribose and uridine administration
- Ketogenic diet
- S-adenosyl-l-methionine
Prognosis
The prognosis of this condition in childhood usually has a stable outcome, whereas in neonatal is almost always fatal, according to Jurecka, et al.[5]
References
- Online Mendelian Inheritance in Man (OMIM): 103050
- "Adenylosuccinase deficiency". rarediseases.info.nih.gov. Genetic and Rare Diseases Information Center (GARD) – an NCATS Program. Retrieved 22 December 2016.
- "Adenylosuccinate lyase deficiency - Conditions - GTR - NCBI". www.ncbi.nlm.nih.gov. Retrieved 22 December 2016.
- RESERVED, INSERM US14 -- ALL RIGHTS. "Orphanet: Adenylosuccinate lyase deficiency". www.orpha.net. Retrieved 22 December 2016.
- Jurecka, Agnieszka; Zikanova, Marie; Kmoch, Stanislav; Tylki-Szymańska, Anna (2014-08-12). "Adenylosuccinate lyase deficiency". Journal of Inherited Metabolic Disease. 38 (2): 231–242. doi:10.1007/s10545-014-9755-y. ISSN 0141-8955. PMC 4341013. PMID 25112391.
- Reference, Genetics Home. "adenylosuccinate lyase deficiency". Genetics Home Reference. Retrieved 18 December 2016.
- Jaeken and Van den Berge, "Adenylosuccinate Lyase Deficiency", The Metabolic and Molecular Bases of Inherited Diseases, Vol. 2, 8th ed., McGraw-Hill; New York, 2001.
- SPIEGEL, E; COLMAN, R; PATTERSON, D (September 2006). "Adenylosuccinate lyase deficiency". Molecular Genetics and Metabolism. 89 (1–2): 19–31. doi:10.1016/j.ymgme.2006.04.018. PMID 16839792. – via ScienceDirect (Subscription may be required or content may be available in libraries.)
- Reference, Genetics Home. "ADSL gene". Genetics Home Reference. Retrieved 21 December 2016.
Further reading
- Saudubray, Jean-Marie; Berghe, Georges van den; Walter, John H. (2011-11-16). Inborn Metabolic Diseases: Diagnosis and Treatment. Springer Science & Business Media. ISBN 9783642157202.
- Blau, Nenad; Hoffmann, Georg F.; Leonard, J. V.; Clarke, Joe T. R. (2006-01-16). Physician's Guide to the Treatment and Follow-Up of Metabolic Diseases. Springer Science & Business Media. ISBN 9783540289623.
External links
| Classification | |
|---|---|
| External resources |