Adenine phosphoribosyltransferase deficiency
Adenine phosphoribosyltransferase deficiency is an autosomal recessive[1] metabolic disorder associated with a mutation in the enzyme adenine phosphoribosyltransferase.[2]
| Adenine phosphoribosyltransferase deficiency | |
|---|---|
| Other names | APRT deficiency or 2,8 Dihydroxyadenine urolithiasis |
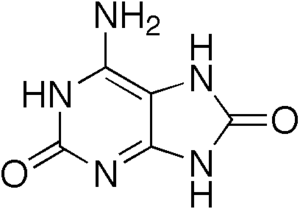 | |
| Dihydroxyadenine, an insoluble purine | |
| Specialty | Endocrinology |
Signs and symptoms
Most patients with APRT deficiency have repeated episodes of kidney stones that are not detected by a conventional x-ray study. However, all stones are easily detected by other medical imaging methods such as ultrasound or computerized tomography (CT) scan. A minority of patients develop symptoms of kidney failure. Kidney stones are often associated with severe loin or abdominal pain. Symptoms associated with kidney failure are largely nonspecific such as increased fatigue and weakness, poor appetite, and weight loss. Children with the disease may have similar symptoms as adults. In young children, APRT deficiency can cause reddish-brown diaper spots.
Genetics
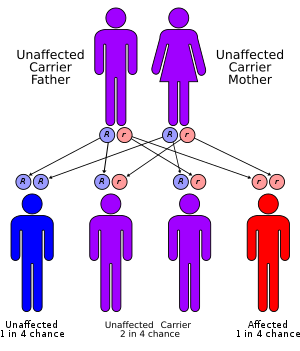
APRT deficiency is inherited in an autosomal recessive manner.[1] This means the defective gene responsible for the disorder is located on an autosome, and two copies of the defective gene (one inherited from each parent) are required in order to be born with the disorder. The parents of an individual with an autosomal recessive disorder both carry one copy of the defective gene, but usually do not experience any signs or symptoms of the disorder.
Characteristics
The disorder results in accumulation of the insoluble purine 2,8-dihydroxyadenine.[3] It can result in nephrolithiasis (kidney stones), acute renal failure and permanent kidney damage.
More than 300 individuals with this disease have been reported world-wide but it is not known how common this medical problem truly is. Patients with the disease deficiency lack the enzyme adenine phosphoribosyltransferase and therefore have difficulties breaking down dietary substances called purines, resulting in accumulation of a compound called 2,8-dihydroxyadenine (2,8-DHA) that is excreted by the kidneys. Up to 70% of affected patients, have red hair or relatives with this hair color.
Diagnosis
Treatment
References
- Kamatani, N (December 1996). "Adenine phosphoribosyltransferase(APRT) deficiency" (Free full text). Nippon Rinsho. Japanese Journal of Clinical Medicine. 54 (12): 3321–7. ISSN 0047-1852. PMID 8976113.
- Terai, C; Hakoda, M; Yamanaka, H; Kamatani, N; Okai, M; Takahashi, F; Kashiwazaki, S (November 1995). "Adenine phosphoribosyltransferase deficiency identified by urinary sediment analysis: cellular and molecular confirmation". Clinical Genetics. 48 (5): 246–50. doi:10.1111/j.1399-0004.1995.tb04098.x. ISSN 0009-9163. PMID 8825602.
- Funato, T; Nishiyama, Y; Ioritani, N; Matsuki, R; Yoshida, K; Kaku, M; Sasaki, T; Ideguchi, H; Ono, J (2000). "Detection of mutations in adenine phosphoribosyltransferase (APRT) deficiency using the LightCycler system". Journal of Clinical Laboratory Analysis. 14 (6): 274–9. doi:10.1002/1098-2825(20001212)14:6<274::AID-JCLA5>3.0.CO;2-2. ISSN 0887-8013. PMC 6808163. PMID 11138609.
External links
| Classification |
|
|---|---|
| External resources |
- Adenine phosphoribosyltransferase deficiency at NIH's Office of Rare Diseases