Superantigen
Superantigens (SAgs) are a class of antigens that result in excessive activation of the immune system. Specifically it causes non-specific activation of T-cells resulting in polyclonal T cell activation and massive cytokine release. SAgs are produced by some pathogenic viruses and bacteria most likely as a defense mechanism against the immune system.[1] Compared to a normal antigen-induced T-cell response where 0.0001-0.001% of the body's T-cells are activated, these SAgs are capable of activating up to 20% of the body's T-cells.[2] Furthermore, Anti-CD3 and Anti-CD28 antibodies (CD28-SuperMAB) have also shown to be highly potent superantigens (and can activate up to 100% of T cells).
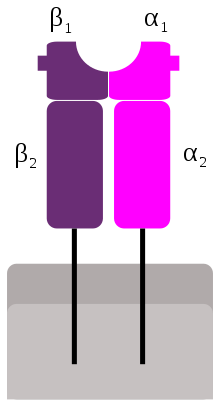
The large number of activated T-cells generates a massive immune response which is not specific to any particular epitope on the SAg thus undermining one of the fundamental strengths of the adaptive immune system, that is, its ability to target antigens with high specificity. More importantly, the large number of activated T-cells secrete large amounts of cytokines, the most important of which is Interferon gamma. This excess amount of IFN-gamma in turn activates the macrophages. The activated macrophages, in turn, over-produce proinflammatory cytokines such as IL-1, IL-6 and TNF-alpha. TNF-alpha is particularly important as a part of the body's inflammatory response. In normal circumstances it is released locally in low levels and helps the immune system defeat pathogens. However, when it is systemically released in the blood and in high levels (due to mass T-cell activation resulting from the SAg binding), it can cause severe and life-threatening symptoms, including shock and multiple organ failure.
Structure
SAgs are produced intracellularly by bacteria and are released upon infection as extracellular mature toxins.[3]
The sequences of these toxins are relatively conserved among the different subgroups. More important than sequence homology, the 3D structure is very similar among different SAgs resulting in similar functional effects among different groups.[4][5]
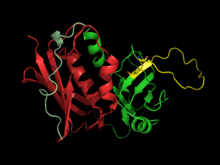
Crystal structures of the enterotoxins reveals that they are compact, ellipsoidal proteins sharing a characteristic two-domain folding pattern comprising an NH2-terminal β barrel globular domain known as the oligosaccharide / oligonucleotide fold, a long α-helix that diagonally spans the center of the molecule, and a COOH terminal globular domain.[4]
The domains have binding regions for the major histocompatibility complex class II (MHC class II) and the T-cell receptor (TCR), respectively.[6]
Binding
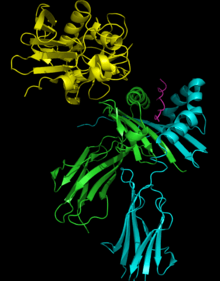
Superantigens bind first to the MHC class II and then coordinate to the variable alpha- or beta chain of T-cell Receptors (TCR)[5][7][8]
MHC Class II
SAgs show preference for the HLA-DQ form of the molecule.[8] Binding to the α-chain puts the SAg in the appropriate position to coordinate to the TCR.
Less commonly, SAgs attach to the polymorphic MHC class II β-chain in an interaction mediated by a zinc ion coordination complex between three SAg residues and a highly conserved region of the HLA-DR β chain.[5] The use of a zinc ion in binding leads to a higher affinity interaction.[4] Several staphylococcal SAgs are capable of cross-linking MHC molecules by binding to both the α and β chains.[4][5] This mechanism stimulates cytokine expression and release in antigen presenting cells as well as inducing the production of costimulatory molecules that allow the cell to bind to and activate T cells more effectively.[5]
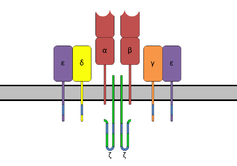
T-cell receptor
T-cell binding region of the SAg interacts with the Variable region on the Beta chain of the T-cell Receptor. A given SAg can activate a large proportion of the T-cell population because the human T-cell repertoire comprises only about 50 types of Vβ elements and some SAgs are capable of binding to multiple types of Vβ regions. This interaction varies slightly among the different groups of SAgs.[6] Variability among different people in the types of T-cell regions that are prevalent explains why some people respond more strongly to certain SAgs. Group I SAgs contact the Vβ at the CDR2 and framework region of the molecule.[9][10] SAgs of Group II interact with the Vβ region using mechanisms that are conformation-dependent. These interactions are for the most part independent of specific Vβ amino acid side-chains. Group IV SAgs have been shown to engage all three CDR loops of certain Vβ forms.[9][10] The interaction takes place in a cleft between the small and large domains of the SAg and allows the SAg to act as a wedge between the TCR and MHC. This displaces the antigenic peptide away from the TCR and circumvents the normal mechanism for T-cell activation.[5][11]
The biological strength of the SAg (its ability to stimulate) is determined by its affinity for the TCR. SAgs with the highest affinity for the TCR elicit the strongest response.[12] SPMEZ-2 is the most potent SAg discovered to date.[12]
T-cell signaling
The SAg cross-links the MHC and the TCR inducing a signaling pathway that results in the proliferation of the cell and production of cytokines. This occurs because a cognate antigen activates a T cell not because of its structure per se, but because its affinity allows it to bind the TCR for a lengthy enough time period, and the SAg mimics this temporal bonding. Low levels of Zap-70 have been found in T-cells activated by SAgs, indicating that the normal signaling pathway of T-cell activation is impaired.[13]
It is hypothesized that Fyn rather than Lck is activated by a tyrosine kinase, leading to the adaptive induction of anergy.[14]
Both the protein kinase C pathway and the protein tyrosine kinase pathways are activated, resulting in upregulating production of proinflammatory cytokines.[15]
This alternative signaling pathway impairs the calcium/calcineurin and Ras/MAPkinase pathways slightly,[14] but allows for a focused inflammatory response.
Direct effects
SAg stimulation of antigen presenting cells and T-cells elicits a response that is mainly inflammatory, focused on the action of Th1 T-helper cells. Some of the major products are IL-1, IL-2, IL-6, TNF-α, gamma interferon (IFN-γ), macrophage inflammatory protein 1α (MIP-1α), MIP-1β, and monocyte chemoattractant protein 1 (MCP-1).[15]
This excessive uncoordinated release of cytokines, (especially TNF-α), overloads the body and results in rashes, fever, and can lead to multi-organ failure, coma and death.[8][10]
Deletion or anergy of activated T-cells follows infection. This results from production of IL-4 and IL-10 from prolonged exposure to the toxin. The IL-4 and IL-10 downregulate production of IFN-gamma, MHC Class II, and costimulatory molecules on the surface of APCs. These effects produce memory cells that are unresponsive to antigen stimulation.[16][17]
One mechanism by which this is possible involves cytokine-mediated suppression of T-cells. MHC crosslinking also activates a signaling pathway that suppresses hematopoiesis and upregulates Fas-mediated apoptosis.[18]
IFN-α is another product of prolonged SAg exposure. This cytokine is closely linked with induction of autoimmunity,[19] and the autoimmune disease Kawasaki disease is known to be caused by SAg infection.[12]
SAg activation in T-cells leads to production of CD40 ligand which activates isotype switching in B cells to IgG and IgM and IgE.[20]
To summarize, the T-cells are stimulated and produce excess amounts of cytokine resulting in cytokine-mediated suppression of T-cells and deletion of the activated cells as the body returns to homeostasis. The toxic effects of the microbe and SAg also damage tissue and organ systems, a condition known as toxic shock syndrome.[20]
If the initial inflammation is survived, the host cells become anergic or are deleted, resulting in a severely compromised immune system.
Superantigenicity independent (indirect) effects
Apart from their mitogenic activity, SAgs are able to cause symptoms that are characteristic of infection.[1]
One such effect is vomiting. This effect is felt in cases of food poisoning, when SAg-producing bacteria release the toxin, which is highly resistant to heat. There is a distinct region of the molecule that is active in inducing gastrointestinal toxicity.[1] This activity is also highly potent, and quantities as small as 20-35 μg of SAg are able to induce vomiting.[8]
SAgs are able to stimulate recruitment of neutrophils to the site of infection in a way that is independent of T-cell stimulation. This effect is due to the ability of SAgs to activate monocytic cells, stimulating the release of the cytokine TNF-α, leading to increased expression of adhesion molecules that recruit leukocytes to infected regions. This causes inflammation in the lungs, intestinal tissue, and any place that the bacteria have colonized.[21] While small amounts of inflammation are natural and helpful, excessive inflammation can lead to tissue destruction.
One of the more dangerous indirect effects of SAg infection concerns the ability of SAgs to augment the effects of endotoxins in the body. This is accomplished by reducing the threshold for endotoxicity. Schlievert demonstrated that, when administered conjunctively, the effects of SAg and endotoxin are magnified as much as 50,000 times.[7] This could be due to the reduced immune system efficiency induced by SAg infection. Aside from the synergistic relationship between endotoxin and SAg, the “double hit” effect of the activity of the endotoxin and the SAg result in effects more deleterious that those seen in a typical bacterial infection. This also implicates SAgs in the progression of sepsis in patients with bacterial infections.[20]
Diseases associated with superantigen production
Treatment
The primary goals of medical treatment are to hemodynamically stabilize the patient and, if present, to eliminate the microbe that is producing the SAgs. This is accomplished through the use of vasopressors, fluid resuscitation and antibiotics.[1]
The body naturally produces antibodies to some SAgs, and this effect can be augmented by stimulating B-cell production of these antibodies.[24]
Immunoglobulin pools are able to neutralize specific antibodies and prevent T-cell activation. Synthetic antibodies and peptides have been created to mimic SAg-binding regions on the MHC class II, blocking the interaction and preventing T cell activation.[1]
Immunosuppressants are also employed to prevent T-cell activation and the release of cytokines. Corticosteroids are used to reduce inflammatory effects.[20]
Evolution of superantigen production
SAg production effectively corrupts the immune response, allowing the microbe secreting the SAg to be carried and transmitted unchecked. One mechanism by which this is done is through inducing anergy of the T-cells to antigens and SAgs.[13][16] Lussow and MacDonald demonstrated this by systematically exposing animals to a streptococcal antigen. They found that exposure to other antigens after SAg infection failed to elicit an immune response.[16] In another experiment, Watson and Lee discovered that memory T-cells created by normal antigen stimulation were anergic to SAg stimulation and that memory T-cells created after a SAg infection were anergic to all antigen stimulation. The mechanism by which this occurred was undetermined.[13] The genes that regulate SAg expression also regulate mechanisms of immune evasion such as M protein and Bacterial capsule expression, supporting the hypothesis that SAg production evolved primarily as a mechanism of immune evasion.[25]
When the structure of individual SAg domains has been compared to other immunoglobulin-binding streptococcal proteins (such as those toxins produced by E. coli) it was found that the domains separately resemble members of these families. This homology suggests that the SAgs evolved through the recombination of two smaller β-strand motifs.[26]
Endogenous SAgs
Minor lymphocyte stimulating (Mls) exotoxins were originally discovered in the thymic stromal cells of mice. These toxins are encoded by SAg genes that were incorporated into the mouse genome from the mouse mammary tumour virus (MMTV). The presence of these genes in the mouse genome allows the mouse to express the antigen in the thymus as a means of negatively selecting for lymphocytes with a variable Beta region that is susceptible to stimulation by the viral SAg. The result is that these mice are immune to infection by the virus later in life.[1]
Similar endogenous SAg-dependent selection has yet to be identified in the human genome, but endogenous SAgs have been discovered and are suspected of playing an integral role in viral infection. Infection by the Epstein–Barr virus, for example, is known to cause production of a SAg in infected cells, yet no gene for the toxin has been found on the genome of the virus. The virus manipulates the infected cell to express its own SAg genes, and this helps it to evade the host immune system. Similar results have been found with rabies, cytomegalovirus, and HIV.[1]
References
- Llewelyn M, Cohen J (March 2002). "Superantigens: microbial agents that corrupt immunity". Lancet Infect Dis. 2 (3): 156–62. doi:10.1016/S1473-3099(02)00222-0. PMID 11944185.
- Li H., Llera A., Malchiodi E.L., Mariuzza R.A. The structural basis of T cell activation by superantigens. Annu. Rev. Immunol. 1999;17:435–466. doi: 10.1146/annurev.immunol.17.1.435.
- Sriskandan S, Faulkner L, Hopkins P (2007). "Streptococcus pyogenes: Insight into the function of the streptococcal superantigens". Int. J. Biochem. Cell Biol. 39 (1): 12–9. doi:10.1016/j.biocel.2006.08.009. PMID 17029999.
- Petersson K, Forsberg G, Walse B (April 2004). "Interplay between superantigens and immunoreceptors". Scand. J. Immunol. 59 (4): 345–55. doi:10.1111/j.0300-9475.2004.01404.x. PMID 15049778.
- Mehindate K, Thibodeau J, Dohlsten M, Kalland T, Sékaly RP, Mourad W (November 1995). "Cross-linking of major histocompatibility complex class II molecules by staphylococcal enterotoxin A superantigen is a requirement for inflammatory cytokine gene expression". J. Exp. Med. 182 (5): 1573–7. doi:10.1084/jem.182.5.1573. PMC 2192187. PMID 7595227.
- Papageorgiou AC, Tranter HS, Acharya KR (March 1998). "Crystal structure of microbial superantigen staphylococcal enterotoxin B at 1.5 A resolution: implications for superantigen recognition by MHC class II molecules and T-cell receptors". J. Mol. Biol. 277 (1): 61–79. doi:10.1006/jmbi.1997.1577. PMID 9514739.
- Schlievert PM (April 1982). "Enhancement of host susceptibility to lethal endotoxin shock by staphylococcal pyrogenic exotoxin type C". Infect. Immun. 36 (1): 123–8. doi:10.1128/IAI.36.1.123-128.1982. PMC 351193. PMID 7042568.
- Alouf JE, Müller-Alouf H (February 2003). "Staphylococcal and streptococcal superantigens: molecular, biological and clinical aspects". Int. J. Med. Microbiol. 292 (7–8): 429–40. doi:10.1078/1438-4221-00232. PMID 12635926.
- Brouillard JN, Günther S, Varma AK, et al. (April 2007). "Crystal structure of the streptococcal superantigen SpeI and functional role of a novel loop domain in T cell activation by group V superantigens". J. Mol. Biol. 367 (4): 925–34. doi:10.1016/j.jmb.2007.01.024. PMID 17303163.
- Buonpane RA, Moza B, Sundberg EJ, Kranz DM (October 2005). "Characterization of T cell receptors engineered for high affinity against toxic shock syndrome toxin-1". J. Mol. Biol. 353 (2): 308–21. doi:10.1016/j.jmb.2005.08.041. PMID 16171815.
- Li H, Llera A, Tsuchiya D, et al. (December 1998). "Three-dimensional structure of the complex between a T cell receptor beta chain and the superantigen staphylococcal enterotoxin B". Immunity. 9 (6): 807–16. doi:10.1016/S1074-7613(00)80646-9. PMID 9881971.
- Arcus VL, Proft T, Sigrell JA, Baker HM, Fraser JD, Baker EN (May 2000). "Conservation and variation in superantigen structure and activity highlighted by the three-dimensional structures of two new superantigens from Streptococcus pyogenes". J. Mol. Biol. 299 (1): 157–68. doi:10.1006/jmbi.2000.3725. PMID 10860729.
- Watson AR, Lee WT (August 2006). "Defective T cell Receptor-mediated Signal Transduction in Memory CD4 T Lymphocytes Exposed to Superantigen or anti-T cell Receptor Antibodies". Cell. Immunol. 242 (2): 80–90. doi:10.1016/j.cellimm.2006.09.008. PMC 1829409. PMID 17083922.
- Choi S, Schwartz RH (June 2007). "Molecular mechanisms for adaptive tolerance and other T cell anergy models". Semin. Immunol. 19 (3): 140–52. doi:10.1016/j.smim.2007.02.005. PMC 2045643. PMID 17400472.
- Stiles BG, Krakauer (2005). "Staphylococcal Enterotoxins: a Purging Experience in Review, Part I". Clinical Microbiology Newsletter. 27 (23): 23. doi:10.1016/j.clinmicnews.2005.11.001.
- Lussow AR, MacDonald HR (February 1994). "Differential effects of superantigen-induced "anergy" on priming and effector stages of a T cell-dependent antibody response". Eur. J. Immunol. 24 (2): 445–9. doi:10.1002/eji.1830240227. PMID 8299694.
- Miller C, Ragheb JA, Schwartz RH (July 1999). "Anergy and Cytokine-Mediated Suppression as Distinct Superantigen-Induced Tolerance Mechanisms in Vivo". J. Exp. Med. 190 (1): 53–64. doi:10.1084/jem.190.1.53. PMC 2195559. PMID 10429670.
- Yamaguchi M, Nadler S, Lee JW, Deeg HJ (September 1999). "Induction of negative regulators of haematopoiesis in human bone marrow cells by HLA-DR cross-linking". Transpl. Immunol. 7 (3): 159–68. doi:10.1016/S0966-3274(99)80035-5. PMID 10608299.
- Stauffer Y, Marguerat S, Meylan F, et al. (October 2001). "Interferon-alpha-induced endogenous superantigen. a model linking environment and autoimmunity". Immunity. 15 (4): 591–601. doi:10.1016/S1074-7613(01)00212-6. PMID 11672541.
- Jabara HH, Geha RS (October 1996). "The superantigen toxic shock syndrome toxin-1 induces CD40 ligand expression and modulates IgE isotype switching". Int. Immunol. 8 (10): 1503–10. doi:10.1093/intimm/8.10.1503. PMID 8921429.
- Diener K, Tessier P, Fraser J, Köntgen F, McColl SR (June 1998). "Induction of acute inflammation in vivo by staphylococcal superantigens I: Leukocyte recruitment occurs independently of T lymphocytes and major histocompatibility complex Class II molecules". Lab. Invest. 78 (6): 647–56. PMID 9645755.
- Van Cauwenberge P, Gevaert P, Van Hoecke H, Van Zele T, Bachert C (2005). "New insights into the pathology of nasal polyposis: the role of superantigens and IgE". Verh K Acad Geneeskd Belg. 67 (5–28): 5–28, discussion 29–32. PMID 15828304.
- Salgado-Pabón W, et al. (2013) Superantigens are critical for Staphylococcus aureus infective endocarditis, sepsis, and acute kidney injury. MBio 4:e00494-00413.
- Erlandsson E, Andersson K, Cavallin A, Nilsson A, et al. (2003). "Identification of the Antigenic Epitopes in Staphylococcal Enterotoxins A and E and Design of a Superantigen for Human Cancer Therapy". J. Mol. Biol. 333 (5): 893–905. doi:10.1016/j.jmb.2003.09.009. PMID 14583188.
- Cleary PP, McLandsborough L, Ikeda L, Cue D, Krawczak J, Lam H (April 1998). "High-frequency intracellular infection and erythrogenic toxin A expression undergo phase variation in M1 group A streptococci". Mol. Microbiol. 28 (1): 157–67. doi:10.1046/j.1365-2958.1998.00786.x. PMID 9593304.
- Bachert C, Gevaert P, van Cauwenberge P (June 2002). "Staphylococcus aureus enterotoxins: a key in airway disease?". Allergy. 57 (6): 480–7. doi:10.1034/j.1398-9995.2002.02156.x. PMID 12028112.
Rasooly, R., Do, P. and Hernlem, B. (2011) Auto-presentation of Staphylococcal enterotoxin A by mouse CD4+ T cells. Open Journal of Immunology, 1, 8-14.
Further reading
- Superantigen Web Database at Birkbeck, University of London
- List of Superantigen Proteins from UniProt
- Superantigens at the US National Library of Medicine Medical Subject Headings (MeSH)
External links
